

Congenital Nail Disorders among Children with Suspected Ectodermal Dysplasias
by
Sigrun Maier-Wohlfart
1,2,*,
Carmen Aicher
1,2,
Ines Willershausen
3,
Nicolai Peschel
1,2,
Udo Meißner
4,
Lina Gölz
3 and
Holm Schneider
1,2,* 1
Center for Ectodermal Dysplasias Erlangen (CEDER), University Hospital Erlangen, 91054 Erlangen, Germany
2
Department of Pediatrics, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91054 Erlangen, Germany
3
Department of Orthodontics and Orofacial Orthopedics, University Hospital Erlangen, 91054 Erlangen, Germany
4
Independent Researcher, 96047 Bamberg, Germany
*
Authors to whom correspondence should be addressed.
Genes 2022, 13(11), 2119; https://doi.org/10.3390/genes13112119
Submission received: 29 September 2022
/
Revised: 26 October 2022
/
Accepted: 8 November 2022
/
Published: 15 November 2022
(This article belongs to the Special Issue Molecular Biology and Treatment of Genodermatoses)
Abstract
:We report on a cohort of 204 children referred between January 2017 and January 2022 to the German Center for Ectodermal Dysplasias, Erlangen. The most frequent reasons for referral were tooth malformations and lack of multiple teeth leading to the suspicion of an ectodermal dysplasia. Many patients also suffered from being unable to perspire. Nail abnormalities, in contrast, represented a much rarer finding, albeit the impact on some individuals was large. As ectodermal dysplasias are congenital genetic conditions affecting the development and/or homeostasis of two or more ectodermal derivatives, including hair, teeth, nails, and certain glands, we analyzed congenital nail disorders detected in these patients. Dystrophic or otherwise abnormal nails were evident in 17 of 18 subjects with pathogenic WNT10A or GJB6 variants but in none of 161 children with EDA variants underlying X-linked hypohidrotic ectodermal dysplasia. However, 2 of 17 children who carry mutations in EDAR or EDARADD, two other genes involved in the ectodysplasin A signaling pathway, showed nail abnormalities, such as brittle or hypoplastic nails. TP63 variants were regularly associated with nail disorders. In one girl, anonychia congenita caused by a compound heterozygous variant of the R-spondin-4 gene (RSPO4) was diagnosed. Thus, nail dysplasia is rarer among patients with ectodermal dysplasia than commonly thought.
1. Introduction
Ectodermal dysplasias are a subgroup of genodermatoses, the entirety of genetic skin diseases, with X-linked, autosomal recessive, or autosomal dominant modes of inheri-tance. The molecular causes of most of them have been identified, elucidating how distinct alterations of signaling pathways can affect the normal embryonic development of ectodermal derivatives like skin, teeth, hair, nails, and certain eccrine glands (e.g., sebaceous, sweat or mammary glands). Although nail abnormalities are considered to be a common feature of many genodermatoses, their prevalence among patients with ectodermal dysplasia (ED) is unknown, as data from large cohorts have been difficult to collect. Furthermore, nail alterations may also be acquired (e.g., related to infectious and inflammatory disorders or trauma) and are often poorly described in the available case series [1,2]. This is in contrast to the fact that, collectively, hereditary, and acquired nail disorders account for 3–11% of pediatric dermatology consultations [3,4]. Congenital nail abnormalities can occur isolated or as a component of genetic syndromes and may involve changes in nail color, texture, and/or shape [5]. In our experience, they do not only facilitate differential diagnosis, but their clinical relevance, particularly for females, is also underestimated.
This paper summarizes the nail-related findings in a large cohort of pediatric patients with clinically suspected ED. X-linked hypohidrotic ectodermal dysplasia (XLHED; Christ–Siemens–Touraine syndrome; MIM #305100), the most frequent form of ED, is characterized by a symptom triad of hypotrichosis, hypo- or even anodontia, and hypo- or anhidrosis, caused by pathogenic variants of the gene EDA (MIM *300451) which encodes the signaling protein ectodysplasin A1. Autosomal recessive and autosomal dominant forms of hypohidrotic ED are often evoked by pathogenic variants of either EDAR (MIM *604095) coding for the ectodysplasin A receptor (EDAR) or EDARADD (MIM *606603) encoding the EDAR-associated death domain adapter. The phenotype may be very similar to that of XLHED due to the common molecular signaling pathway [6,7,8]. Patients with Incontinentia pigmenti (IP; MIM #308300), a rare X-linked disorder caused by pathogenic variants of the gene IKBKG (MIM *300248) display a characteristic skin blistering during early infancy followed by hyperpigmentation, tooth anomalies, sparse hair, and sometimes severe involvement of the eyes and the central nervous system [9]. In hemizygous males, IP is associated with severe immunodeficiency which is usually lethal. Pathogenic WNT10A (MIM *606268) variants can be responsible for isolated tooth agenesis as well as for odonto-onycho-dermal dysplasia (OODD; MIM #257980) and Schöpf–Schulz–Passarge syndrome (SSPS; MIM #224750). Phenotypic features of recessively inherited OODD and SSPS include hypodontia, nail dystrophy, and palmoplantar hyperkeratosis [10,11,12]. Pathogenic variants of GJB6 (MIM *604418) underlie Clouston syndrome (MIM #129500), an autosomal dominant disorder with the key symptoms nail dystrophy, alopecia, and palmoplantar hyperkeratosis [13].
Moreover, there are complex ED syndromes, such as ankyloblepharon-ectodermal dysplasia-cleft lip/palate (AEC) syndrome (MIM #106260) and ectrodactyly-ectodermal dysplasia-cleft lip/palate (EEC) syndrome (MIM #604292), two dominantly inherited disorders caused by pathogenic variants of the gene TP63 (MIM *603273) that encodes a key transcription factor, tumor protein 63 (p63). The clinical findings in patients with p63-related syndromes include alopecia, dystrophic nails, hypodontia, and (often bilateral) cleft lip and/or palate. In addition, AEC syndrome is characterized by congenital anomalies of the eyelids, while ectrodactyly of hands and feet is a specific feature of EEC syndrome [14,15]. Relevant hypohidrosis does not belong to the regular symptoms associated with TP63 mutations [16].
The absence or maldevelopment of finger- and toenails is the main characteristic of anonychia congenita and its milder phenotypic variant, nonsyndromic congenital nail disorder-4 (NDNC4; MIM #206800). Both are autosomal recessive disorders due to pathogenic variants of RSPO4 (MIM *610573) encoding roof plate-specific (R)-spondin 4. They show a variable phenotype, ranging from the complete absence of the nail field to one of reduced size, with or without a rudimentary nail [17,18,19]. The R-spondin 4 protein is expressed in the nail mesenchyme and acts as an activator of the Wnt/β-catenin signaling pathway [20,21].
Continuous referral of patients with all forms of ED to our center and their systematic clinical examination allowed a retrospective compilation of nail disorders seen during the last five years.
2. Subjects and Methods
2.1. Patients and Study Design
A cohort of 204 children (both sexes) referred between January 2017 and January 2022 with clinically suspected ectodermal dysplasia to the German Center for Ectodermal Dysplasias Erlangen (CEDER) was investigated. The inclusion criterion was the presence of abnormalities affecting at least two different ectodermal structures. Those patients who had not yet obtained a molecular genetic diagnosis were enrolled in the study “Detection and functional investigation of potentially pathogenic variants of HED-associated genes and new candidate regions”, approved by the Ethics Committee of the Friedrich-Alexander-Universität Erlangen-Nürnberg. Patients referred because of tooth malformations and lack of multiple teeth were usually seen in a joined pediatric and dental clinic. In all patients and in some of their family members if indicated, the nail phenotype (size, shape, color, fragility, thickness, signs of onycholysis) was assessed, focusing on congenital nail defects. Localized abnormalities that did not affect the majority of finger- or toenails were not considered. Nail disorders were documented during the first presentation at our center by photographs of both hands and feet. The study participants and/or their legal guardians provided written informed consent to the use of DNA for genetic analysis and further research.
2.2. DNA Analysis
Standard gene variant analysis (DNA isolation from blood, polymerase chain reaction, subsequent Sanger sequencing, or multiplex ligation-dependent probe amplification) was performed as described previously [22]. Oligonucleotide primer sequences and thermal cycling conditions are available upon request. The software program ChromasPro 2.1.9 (Technelysium Pty Ltd., South Brisbane, Australia) was used for analysis of the electropherograms. In two cases, samples were sent to a provider of next-generation sequencing services (CeGaT GmbH, Tübingen, Germany) for whole-exome sequencing (WES) using the Illumina NovaSeq6000 Sequencing Systems. Datasets were analyzed bio-informatically with the Golden Helix GenomeBrowse tool (Golden Helix, Bozeman, MT, USA). Each detected variant was assessed with the mutation prediction tools Mutation Taster (Charité, Berlin, Germany; Cardiff University, Cardiff, UK) and/or the Ensembl Variant Effect Predictor also containing SIFT and PolyPhen-2 scores for protein changes (European Molecular Biology Laboratory’s European Bioinformatics Institute, Hinxton, UK).
2.3. In Silico Protein Structure Analysis
For structure prediction of pathogenic variants of the RSPO4 protein (UniProt: Q2I0M5), three-dimensional model data from AlphaFold (https://α-fold.ebi.ac.uk/ accessed on 16 April 2022) were analyzed with AlphaFold Colab (https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb accessed on 16 April 2022; [23]). UCSF ChimeraX was used for the visualization of the molecular graphics (resource for biocomputing, visualization, and informatics at the University of California [24]).
3. Results
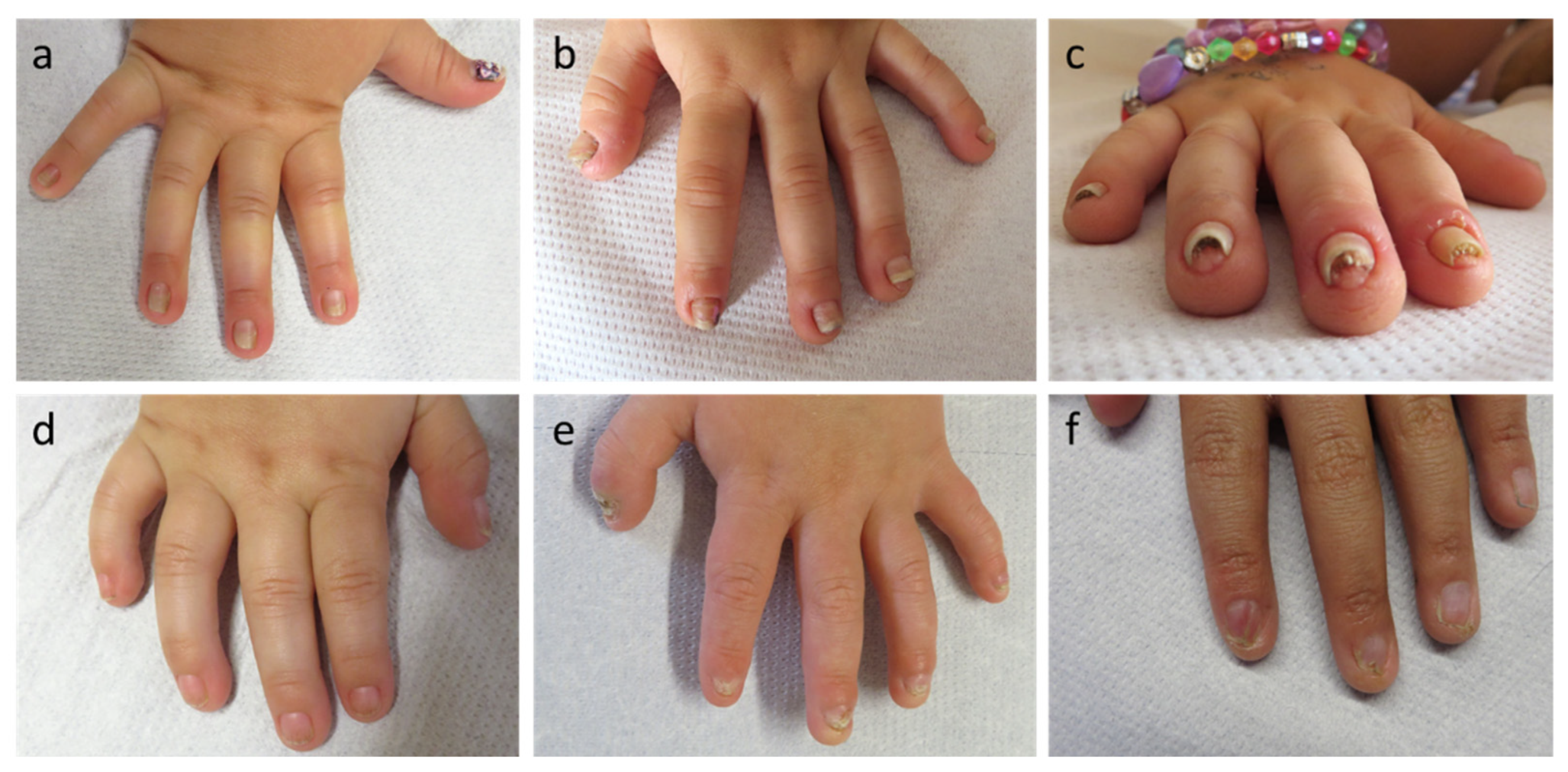
A total of 26 (12.8%) of 204 children with clinically suspected ED had nail abnormalities, such as brittle, hypoplastic or completely missing finger- and/or toenails, at the time of first presentation at our center (Table 1). The nails were regularly affected only in patients with Clouston syndrome (two females; Figure 1a–c), AEC syndrome (one male and three females; Figure 1d–f) and NDNC4 (one female). Approximately 30% of all patients did not yet have a molecular diagnosis of their condition and required molecular genetic analysis. Most individuals with pathogenic WNT10A variants underlying OODD or SSPS displayed characteristic nail dystrophies (six males and nine females; 94%), one infant with numerous missing teeth and moderate skin issues had normal nails. EEC syndrome was associated with nail abnormalities in two out of three male patients. However, not a single one of 161 male or female patients with XLHED and none of the three girls with IP showed obvious peculiarities of their finger- and toenails, while two female patients with autosomal recessive or dominant hypohidrotic ED caused by EDAR and EDARADD mutations, respectively, presented with dystrophic toenails (Table 1).
In cases of Clouston syndrome, typical nail plate malformations included a narrow and dysmorphic shaping, hyperconvexity, milky white discoloration, and thickening of the nails with distal onycholysis (Figure 1a–c). Nail plates of the patients with AEC or EEC syndrome were partially discolored, fragile, frayed at the distal edges and showed ptery-gium formation and underdeveloped cuticles (Figure 1d–f).
One child later diagnosed with anonychia congenita, a two-year-old girl born to non-consanguineous and phenotypically unremarkable parents, was referred to the CEDER because of missing finger- and toenail development. Absence of all nail plates had been observed from birth, but did not relevantly affect grasping or other functions of the hands. Nail bed, matrix, and fold and the surrounding skin seemed to be unaffected and had a healthy appearance (Figure 2a,d). Except for hypoplastic nail plates on the big toes, the mother showed no nail abnormalities (Figure 2b,e). Four toes of the father’s right foot displayed somewhat brittle nails with a trauma-induced subungual hematoma on the second toe (after running a marathon; Figure 2f), whereas the other finger- and toenails were unremarkable (Figure 2c,f). The infant was able to sweat normally, but had rather sparse eyebrows.
Whole-exome sequencing of leukocyte-derived DNA from the index patient was performed. Analysis of the obtained bioinformatic data, particularly with regard to potential pathogenicity of detected variants and known associations of candidate genes with the patient’s phenotype, revealed few regions of interest. Two heterozygous and (probably coincidentally) adjacent alterations in the gene RSPO4, the variant c.296T>C (p.Phe99Ser) and the in-frame deletion c.273_275delTGG (p.Cys91_Gly92delins-Trp), turned out to be the most relevant changes in the exome of the affected girl (Figure 3a).
While the variant RSPO4 c.296T>C is novel and predicted to be disease-causing, a report on RSPO4 c.273_275delTGG has already been included in the Exome Aggregation Consortium (ExAC) database for heterozygous carriers. Variant effect prediction tools classify it as “likely polymorphic”. We assume that both variants add up to cause the infant’s phenotype in a compound-heterozygous manner. Sanger sequencing of genomic DNA of the parents revealed the mother to carry the deletion and the father to be a carrier of the missense variant, each in a heterozygous state (Figure 3b). The variant p.Cys91_Gly92delinsTrp is predicted to disrupt the normal disulfide bond formation bet-ween amino acids 91 and 98 of the wild-type protein (Figure 3c). The phenylalanine at position 99 is highly conserved and neighboring p.Ser100, an amino acid known to be important for the regulation of the Wnt/β-catenin signaling pathway due to its interaction with the leucine-rich repeat-containing G-protein-coupled receptors (LGR) 4, 5, and 6 [25].
In addition, the index patient was compound-heterozygous for two variants of the related gene LGR5 (c.1997T>C/p.Val666Ala and c.2125C>G/p.Pro709Ala) which might also play a role in combining anonychia congenita with a mild hair phenotype.
4. Discussion
Nail disorders may have various phenotypic presentations, ranging from harmless discolorations to complete absence of all nails, as well as different causes, including genetic conditions, injuries, malnutrition or infections [26]. Depending on the severity, they impact the quality of life by causing psychological and/or functional problems [27]. Nail deformities and/or dystrophies are well known features of some ED entities, such as OODD/SSPS, Clouston syndrome, and AEC/EEC syndrome, which was confirmed by our findings. The nail phenotype may be rather specific like in patients with Clouston syndrome or show a wider spectrum of nail plate abnormalities as observed in children with WNT10A or TP63 mutations. In cases of XLHED/HED, diverging information is available in the literature. Some authors stated that nail anomalies are generally not seen in subjects with XLHED, others report about 50% of their patients being affected [28,29,30]. This discrepancy could possibly be explained by subjective definitions of nail defects, especially in mild cases [31]. The complete absence of obvious nail abnormalities in our large cohort of pediatric patients with XLHED, however, indicates that this ED entity does not frequently involve the nails, at least in children. Nail defects become evident with age in some cases, but are usually not severe. Raising awareness and knowledge among patients and physicians is likely to improve the documentation of nail-related issues and will, therefore, lead to more standardized and comparable assessments in future studies.
The prevalence of pure nail disorders is probably underestimated, as patients do not always seek medical advice and abnormalities can easily be missed by physicians. Rare conditions of skin appendages have received relatively little scientific attention so far and no therapies are available for the hereditary forms of isolated nail disorders [32]. Pathogenic variants of five genes (RSPO4, FZD6, HPGD, PLCD1, and COL7A1) have been reported to cause congenital pure nail disorders, among which FZD6 (frizzled class receptor 6) and RSPO4 are responsible for most of the cases [18,20,33,34]. Since hair is usually not affected by such mutations, the patient with anonychia congenita described in this paper was included in the study because of his sparse eyebrows in addition to the missing nails. RSPO4 functions as an agonist binding to the Frizzled receptor family which is involved in the activation of the Wnt/β-catenin signaling pathway. This well-known pathway plays a crucial role in the development of various ectodermal derivatives. LGR4–6 serve as receptors for RSPO proteins and have been assumed to connect with a Frizzled/low-density lipoprotein receptor-related protein co-receptor complex [34,35]. Mediation of the Wnt/β-catenin signaling pathway by RSPO4 seems to be LGR-dependent [25]. With this in mind, the two additional variants of the gene LGR5 found in the patient with RPSO4-associated anonychia congenita are certainly very interesting, although a potential pathomechanism remains elusive.
Although the genetic causes of some isolated nail disorders have not yet been identified, the increasing usage of next-generation sequencing in molecular diagnostics is likely to enable the detection of pathogenic variants of other relevant genes within the next few years [32]. The same applies to complex genetic conditions, such as the ectodermal dysplasias.
5. Conclusions
Nail abnormalities seem to be accessory, sometimes also severe symptoms in the group of patients with pathogenic variants of the genes WNT10A, GJB6, TP63, and RSPO4, but appear to play a minor role in autosomal recessive or dominant forms of HED. In our large cohort of pediatric patients, EDA variants underlying XLHED were not associated with obvious congenital nail disorders. Physicians working in the field of pediatric dermatology as well as dentists and clinical geneticists should be sensitized to this difference, as nail-related findings—similar to disease-specific schemes of missing teeth—may facilitate or even guide the differential diagnosis.
Author Contributions
S.M.-W. and H.S. conceptualized the research. C.A., I.W., U.M. and H.S. performed clinical examinations. S.M.-W. and N.P. conducted experiments and analyzed the molecular genetic data, S.M.-W., L.G. and H.S. curated data. S.M.-W. wrote the first draft of the manu-script and H.S. reviewed and edited it. All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge financial support by Deutsche Forschungsgemeinschaft and Friedrich-Alexander-Universität Erlangen-Nürnberg within the funding program “Open Access Publication Funding”.
Institutional Review Board Statement
The study was approved by the ethics committee of the Friedrich-Alexander-Universität Erlangen-Nürnberg and conducted in accordance with the principles of the declaration of Helsinki.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the patients and their families for participating in this research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Piraccini, B.M.; Starace, M. Nail disorders in infants and children. Curr. Opin. Pediatr. 2014, 26, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Starace, M.; Alessandrini, A.; Piraccini, B.M. Nail disorders in children. Skin Appendage Disord. 2018, 4, 217–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias, A.; Tamayo, L.; Sosa-de-Martínez, C.; Durán-McKinster, C.; Orozco-Covarrubias, L.; Ruiz-Maldonado, R. Prevalence and nature of nail alterations in pediatric patients. Pediatr. Dermatol. 2001, 18, 107–109. [Google Scholar] [CrossRef]
- Tasia, M.; Lecerf, P.; Richert, B.; André, J. Paediatric nail consultation in an academic centre in Belgium: A 10-year retrospective study. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1800–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzberg, A.J. Nail manifestations of systemic diseases. Clin. Podiatr. Med. Surg. 1995, 12, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Kere, J.; Srivastava, A.K.; Montonen, O.; Zonana, J.; Thomas, N.; Ferguson, B.; Munoz, F.; Morgan, D.; Clarke, A.; Baybayan, P.; et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat. Genet. 1996, 13, 409–416. [Google Scholar] [CrossRef]
- Monreal, A.W.; Ferguson, B.M.; Headon, D.J.; Street, S.L.; Overbeek, P.A.; Zonana, J. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat. Genet. 1999, 22, 366–369. [Google Scholar] [CrossRef]
- Kohn, L.L.; Braun, M.; Cordoro, K.M.; McCalmont, T.H.; Shah, S.D.; Frieden, I.J.; Mathur, A.N. Skin and mucosal manifestations in NEMO syndrome: A case series and literature review. Pediatr. Dermatol. 2022, 39, 84–90. [Google Scholar] [CrossRef]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P.; et al. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [CrossRef]
- Krøigård, A.B.; Clemmensen, O.; Gjørup, H.; Hertz, J.M.; Bygum, A. Odonto-onycho-dermal dysplasia in a patient homozygous for a WNT10A nonsense mutation and mild manifestations of ectodermal dysplasia in carriers of the mutation. BMC Dermatol. 2016, 16, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, T.C.; Lee, J.Y.; Hsu, M.M.; Chao, S.C. Case report of Schöpf-Schulz-Passarge syndrome resulting from a missense mutation, p.Arg104Cys, in WNT10A. J. Dermatol. 2018, 45, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Kibar, Z.; Der Kaloustian, V.M.; Brais, B.; Hani, V.; Fraser, F.C.; Rouleau, G.A. The gene responsible for Clouston hidrotic ectodermal dysplasia maps to the pericentromeric region of chromosome 13q. Hum. Mol. Genet. 1996, 5, 543–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, S.M.; de Jong, T.P.; Buss, P.; Hennekam, R.C. EEC syndrome and genitourinary anomalies: An update. Am. J. Med. Genet. 1996, 63, 472–478. [Google Scholar] [CrossRef]
- McGrath, J.A.; Duijf, P.H.; Doetsch, V.; Irvine, A.D.; de Waal, R.; Vanmolkot, K.R.; Wessagowit, V.; Kelly, A.; Atherton, D.J.; Griffiths, W.A.; et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum. Mol. Genet. 2001, 10, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Ferstl, P.; Wohlfart, S.; Schneider, H. Sweating ability of patients with p63-associated syndromes. Eur. J. Pediatr. 2018, 177, 1727–1731. [Google Scholar] [CrossRef]
- Littman, A.; Levin, S. Anonychia as a recessive autosomal trait in man. J. Investig. Dermatol. 1964, 42, 177–178. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, C.; Senderek, J.; Anhuf, D.; Thiel, C.T.; Ekici, A.B.; Poblete-Gutierrez, P.; van Steensel, M.; Seelow, D.; Nürnberg, G.; Schild, H.H.; et al. Mutations in the gene encoding the Wnt-signaling component R-spondin 4 (RSPO4) cause autosomal recessive anonychia. Am. J. Hum. Genet. 2006, 79, 1105–1109. [Google Scholar] [CrossRef] [Green Version]
- Brüchle, N.O.; Frank, J.; Frank, V.; Senderek, J.; Akar, A.; Koc, E.; Rigopoulos, D.; van Steensel, M.; Zerres, K.; Bergmann, C. RSPO4 is the major gene in autosomal-recessive anonychia and mutations cluster in the furin-like cysteine-rich domains of the Wnt signaling ligand R-spondin 4. J. Investig. Dermatol. 2008, 128, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Blaydon, D.C.; Ishii, Y.; O’Toole, E.A.; Unsworth, H.C.; Teh, M.T.; Rüschendorf, F.; Sinclair, C.; Hopsu-Havu, V.K.; Tidman, N.; Moss, C.; et al. The gene encoding R-spondin 4 (RSPO4), a secreted protein implicated in Wnt signaling, is mutated in inherited anonychia. Nat. Genet. 2006, 38, 1245–1247. [Google Scholar] [CrossRef]
- Hsu, C.K.; Romano, M.T.; Nanda, A.; Rashidghamat, E.; Lee, J.Y.W.; Huang, H.Y.; Songsantiphap, C.; Lee, J.Y.; Al-Ajmi, H.; Betz, R.C.; et al. Congenital anonychia and uncombable hair syndrome: Coinheritance of homozygous mutations in RSPO4 and PADI3. J. Investig. Dermatol. 2017, 137, 1176–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohlfart, S.; Hammersen, J.; Schneider, H. Mutational spectrum in 101 patients with hypohidrotic ectodermal dysplasia and breakpoint mapping in independent cases of rare genomic rearrangements. J. Hum. Genet. 2016, 61, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Raza, S.I.; Navid, A.K.; Noor, Z.; Shah, K.; Dar, N.R.; Ahmad, W.; Rashid, S. Gly67Arg substitution in RSPO4 disrupts the WNT signaling pathway due to an abnormal binding pattern with LGRs leading to anonychia. RSC Adv. 2017, 7, 17357–17366. [Google Scholar] [CrossRef] [Green Version]
- Bloom, A.; Blanken, B.; Schlakman, B.; Arena, T.; Mironov, Z.; Vlahovic, T.C. A review of nail dystrophies for the practitioner. Adv. Skin Wound Care 2020, 33, 20–26. [Google Scholar] [CrossRef]
- Belyayeva, E.; Gregoriou, S.; Chalikias, J.; Kontochristopoulos, G.; Koumantaki, E.; Makris, M.; Koti, I.; Katoulis, A.; Katsambas, A.; Rigopoulos, D. The impact of nail disorders on quality of life. Eur. J. Dermatol. 2013, 23, 366–371. [Google Scholar] [CrossRef]
- Nakata, M.; Koshiba, H.; Eto, K.; Nance, W.E. A genetic study of anodontia in X-linked hypohidrotic ectodermal dysplasia. Am. J. Hum. Genet. 1980, 32, 908–919. [Google Scholar]
- Mortier, K.; Wackens, G. Ectodermal dysplasia anhidrotic. Orphanet Encycl. 2004, 3, 1–6. [Google Scholar]
- Fete, M.; Hermann, J.; Behrens, J.; Huttner, K.M. X-linked hypohidrotic ectodermal dysplasia (XLHED): Clinical and diagnostic insights from an international patient registry. Am. J. Med. Genet. Part A 2014, 164, 2437–2442. [Google Scholar] [CrossRef]
- Bellet, J.S. Pediatric nail disorders. Dermatol. Clin. 2021, 39, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Betz, R.C. Nails—More than just an ectodermal appendage: The genetics behind isolated nail disorders. Br. J. Dermatol. 2015, 173, 886. [Google Scholar] [CrossRef] [PubMed]
- Fröjmark, A.S.; Schuster, J.; Sobol, M.; Entesarian, M.; Kilander, M.B.C.; Gabrikova, D.; Nawaz, S.; Baig, S.M.; Schulte, G.; Klar, J.; et al. Mutations in Frizzled 6 cause isolated autosomal-recessive nail dysplasia. Am. J. Hum. Genet. 2011, 88, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Basit, S.; Habib, R.; Kamal, A.; Muhammad, N.; Ahmad, W. Genetics of human isolated hereditary nail disorders. Br. J. Dermatol. 2015, 173, 922–929. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef]
Figure 1.
Exemplary nail abnormalities observed in the patient cohort. (a–c) Dystrophic nails of patients with Clouston syndrome; (d–f) Spectrum of nail plate abnormalities in p63-associated syndromes.
Figure 1.
Exemplary nail abnormalities observed in the patient cohort. (a–c) Dystrophic nails of patients with Clouston syndrome; (d–f) Spectrum of nail plate abnormalities in p63-associated syndromes.

Figure 2.
Phenotypic findings in the case of anonychia congenita (a–c) Fingernails of the index patient, her mother and her father; (d–f) Toenails of the index patient, her mother and her father.
Figure 2.
Phenotypic findings in the case of anonychia congenita (a–c) Fingernails of the index patient, her mother and her father; (d–f) Toenails of the index patient, her mother and her father.

Figure 3.
Molecular genetics of the (familial) case of anonychia congenita. (a) Golden Helix GenomeBrowse analysis of the WES results showing the two probably pathogenic RSPO4 variants c.296T>C and c.273_275delTGG (reverse complement DNA sequence) of the compound-heterozygous index patient; (b) Chromatograms of the parents’ Sanger sequencing results displaying the heterozygous carrier status of mother and father for RSPO4 c.273_275delTGG and c.296T>C, respectively; (c) Predicted three-dimensional structures of wild-type RSPO4, mutant p.Cys91_Gly92delinsTrp and p.Phe99Ser proteins.
Figure 3.
Molecular genetics of the (familial) case of anonychia congenita. (a) Golden Helix GenomeBrowse analysis of the WES results showing the two probably pathogenic RSPO4 variants c.296T>C and c.273_275delTGG (reverse complement DNA sequence) of the compound-heterozygous index patient; (b) Chromatograms of the parents’ Sanger sequencing results displaying the heterozygous carrier status of mother and father for RSPO4 c.273_275delTGG and c.296T>C, respectively; (c) Predicted three-dimensional structures of wild-type RSPO4, mutant p.Cys91_Gly92delinsTrp and p.Phe99Ser proteins.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Proportion of nail abnormalities among children with clinically suspected ED seen at the CEDER in the last five years.
Table 1.
Proportion of nail abnormalities among children with clinically suspected ED seen at the CEDER in the last five years.
| Disease(s) | Affected Gene(s) | Number of Patients Seen 2017–2022 | Patients with Nail Abnormalities |
|---|---|---|---|
| Hypohidrotic ED, X-linked | EDA | 161 | 0 (0%) |
| Hypohidrotic ED, autosomal dominant or recessive | EDAR, EDARADD | 14 | 2 (14.3%) |
| Incontinentia pigmenti | IKBKG | 3 | 0 (0%) |
| Odonto-onycho-dermal dysplasia/ Schöpf-Schulz-Passarge syndrome | WNT10A | 16 | 15 (93.8%) |
| Clouston syndrome | GJB6 2 | 2 | 2 (100%) |
| Ankyloblepharon-ED-cleft lip/palate (AEC) syndrome | TP63 | 4 | 4 (100%) |
| Ectrodactyly-ED-cleft lip/palate (EEC) syndrome | TP63 | 3 | 2 (66.7%) |
| Anonychia congenita | RSPO4 | 1 | 1 (100%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Maier-Wohlfart, S.; Aicher, C.; Willershausen, I.; Peschel, N.; Meißner, U.; Gölz, L.; Schneider, H. Congenital Nail Disorders among Children with Suspected Ectodermal Dysplasias. Genes 2022, 13, 2119. https://doi.org/10.3390/genes13112119
AMA Style
Maier-Wohlfart S, Aicher C, Willershausen I, Peschel N, Meißner U, Gölz L, Schneider H. Congenital Nail Disorders among Children with Suspected Ectodermal Dysplasias. Genes. 2022; 13(11):2119. https://doi.org/10.3390/genes13112119
Chicago/Turabian StyleMaier-Wohlfart, Sigrun, Carmen Aicher, Ines Willershausen, Nicolai Peschel, Udo Meißner, Lina Gölz, and Holm Schneider. 2022. "Congenital Nail Disorders among Children with Suspected Ectodermal Dysplasias" Genes 13, no. 11: 2119. https://doi.org/10.3390/genes13112119
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.