WO2016109795A1 - Deuterated funapide and difluorofunapide - Google Patents
Deuterated funapide and difluorofunapide Download PDFInfo
- Publication number
- WO2016109795A1 WO2016109795A1 PCT/US2015/068276 US2015068276W WO2016109795A1 WO 2016109795 A1 WO2016109795 A1 WO 2016109795A1 US 2015068276 W US2015068276 W US 2015068276W WO 2016109795 A1 WO2016109795 A1 WO 2016109795A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pain
- compound
- deuterium
- same
- compounds
- Prior art date
Links
- 0 *C1(*)Oc(c(*)c(c2c3*=C4N(C(c5ccccc5)c5ccccc5)c5c(*)c(*)c(*)c(*)c5C24)OCc2ccccc2)c3O1 Chemical compound *C1(*)Oc(c(*)c(c2c3*=C4N(C(c5ccccc5)c5ccccc5)c5c(*)c(*)c(*)c(*)c5C24)OCc2ccccc2)c3O1 0.000 description 7
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/20—Spiro-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
Definitions
- ADME absorption, distribution, metabolism and/or excretion
- ADME limitation that affects many medicines is the formation of toxic or biologically reactive metabolites.
- some patients receiving the drug may experience toxicities, or the safe dosing of such drugs may be limited such that patients receive a suboptimal amount of the active agent.
- modifying dosing intervals or formulation approaches can help to reduce clinical adverse effects, but often the formation of such undesirable metabolites is intrinsic to the metabolism of the compound.
- a metabolic inhibitor will be co-administered with a drug that is cleared too rapidly.
- a drug that is cleared too rapidly.
- the FDA recommends that these drugs be co-dosed with ritonavir, an inhibitor of cytochrome P450 enzyme 3 A4 (CYP3 A4), the enzyme typically responsible for their metabolism (see Kempf, D.J. et al., Antimicrobial agents and chemotherapy, 1997, 41(3): 654-60).
- Ritonavir causes adverse effects and adds to the pill burden for HIV patients who must already take a combination of different drugs.
- the CYP2D6 inhibitor quinidine has been added to dextromethorphan for the purpose of reducing rapid CYP2D6 metabolism of dextromethorphan in a treatment of pseudobulbar affect.
- Quinidine has unwanted side effects that greatly limit its use in potential combination therapy (see Wang, L et al., Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt 1): 659-67; and FDA label for quinidine at www.accessdata.fda.gov).
- a potentially attractive strategy for improving a drug's metabolic properties is deuterium modification.
- Deuterium is a safe, stable, nonradioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the ADME properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability.
- the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
- This invention relates to novel, deuterated spiro-oxindole compounds, and pharmaceutically acceptable salts thereof.
- This invention also provides compositions comprising a compound of this invention and the use of such compositions in methods of reducing or eliminating pain and in treating diseases and conditions that are characterized by pain.
- Funapide is currently in Phase II human clinical trials for postherpetic neuralgia, neuropathic pain, osteoarthritic pain, dental pain and erythromelalgia.
- treat means decrease, suppress, attenuate, diminish, arrest, or stabilize the development or progression of a disease (e.g., a disease or disorder delineated herein), lessen the severity of the disease or improve the symptoms associated with the disease.
- Disease means any condition or disorder that damages or interferes with the normal function of a cell, tissue, or organ.
- any atom not specifically designated as a particular isotope is meant to represent any stable isotope of that atom.
- a position is designated specifically as “H” or “hydrogen”
- the position is understood to have hydrogen at its natural abundance isotopic composition.
- a position is designated specifically as “D” or “deuterium”
- the position is understood to have deuterium at an abundance that is at least 3340 times greater than the natural abundance of deuterium, which is 0.015% (i.e., at least 50.1% incorporation of deuterium).
- isotopic enrichment factor means the ratio between the isotopic abundance and the natural abundance of a specified isotope.
- a compound of this invention has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium
- incorporation at each designated deuterium atom at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5%) deuterium incorporation).
- isotopologue refers to a species in which the chemical structure differs from a specific compound of this invention only in the isotopic composition thereof.
- compound when referring to a compound of this invention, refers to a collection of molecules having an identical chemical structure, except that there may be isotopic variation among the constituent atoms of the molecules.
- the relative amount of such isotopologues in a compound of this invention will depend upon a number of factors including the isotopic purity of deuterated reagents used to make the compound and the efficiency of incorporation of deuterium in the various synthesis steps used to prepare the compound. However, as set forth above the relative amount of such isotopologues in toto will be less than 49.9% of the compound. In other embodiments, the relative amount of such isotopologues in toto will be less than 47.5%, less than 40%, less than 32.5%, less than 25%, less than 17.5%, less than 10%, less than 5%, less than 3%, less than 1%, or less than 0.5% of the compound.
- the invention also provides salts of the compounds of the invention.
- a salt of a compound of this invention is formed between an acid and a basic group of the compound, such as an amino functional group, or a base and an acidic group of the compound, such as a carboxyl functional group.
- the compound is a pharmaceutically acceptable acid addition salt.
- pharmaceutically acceptable refers to a component that is, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and other mammals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salt means any non-toxic salt that, upon administration to a recipient, is capable of providing, either directly or indirectly, a compound of this invention.
- pharmaceutically acceptable counterion is an ionic portion of a salt that is not toxic when released from the salt upon administration to a recipient.
- Acids commonly employed to form pharmaceutically acceptable salts include inorganic acids such as hydrogen bisulfide, hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid and phosphoric acid, as well as organic acids such as para- toluenesulfonic acid, salicylic acid, tartaric acid, bitartaric acid, ascorbic acid, maleic acid, besylic acid, fumaric acid, gluconic acid, glucuronic acid, formic acid, glutamic acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, lactic acid, oxalic acid, para-bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid and acetic acid, as well as related inorganic and organic acids.
- organic acids such as para- toluenesulfonic acid, salicylic acid, tartaric acid, bitartaric acid, ascorbic acid
- salts thus include sulfate, pyrosulfate, bisulfate, sulfite, bisulfite, phosphate, monohydrogenphosphate, dihydrogenphosphate, metaphosphate, pyrophosphate, chloride, bromide, iodide, acetate, propionate, decanoate, caprylate, acrylate, formate, isobutyrate, caprate, heptanoate, propiolate, oxalate, malonate, succinate, suberate, sebacate, fumarate, maleate, butyne-l,4-dioate, hexyne-l,6-dioate, benzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, phthalate, terephthalate, sulfonate, xylene sulfonate, phenyl acetate, phenylpropyl
- the compounds of the present invention may contain an asymmetric carbon atom, for example, as the result of deuterium substitution or otherwise. As indicated herein, such stereocenters may be indicated by an asterisk ("*"). As such, compounds of this invention can exist as either individual enantiomers, or mixtures of the two enantiomers. Accordingly, a compound of the present invention may exist as either a racemic mixture or a scalemic mixture, or as individual respective stereoisomers that are substantially free from another possible stereoisomer.
- substantially free of other stereoisomers means less than 25% of other stereoisomers, preferably less than 10% of other stereoisomers, more preferably less than 5% of other stereoisomers and most preferably less than 2% of other stereoisomers are present.
- stable compounds refers to compounds which possess stability sufficient to allow for their manufacture and which maintain the integrity of the compound for a sufficient period of time to be useful for the purposes detailed herein (e.g., formulation into therapeutic products, intermediates for use in production of therapeutic compounds, isolatable or storable intermediate compounds, treating a disease or condition responsive to therapeutic agents).
- Substituted with deuterium refers to the replacement of one or more hydrogen atoms with a corresponding number of deuterium atoms.
- variable may be referred to generally (e.g., "each Y 3 ”) or may be referred to specifically (e.g., Y 3a , Y 3b , Y 3c , etc.). Unless otherwise indicated, when a variable is referred to generally, it is meant to include all specific embodiments of that particular variable.
- each of Y la and Y lb is independently selected from hydrogen, deuterium and fluorine: each Y 2 , Y 3 , Y 4 , Y 5 and Y 6 is independently selected from hydrogen and deuterium;
- Y la , Y lb , Y 2a , Y 2b , Y 3a , Y 3b , Y 3c , Y 3d , Y 4a , Y 4b , Y 5a , Y 5b , Y 6a , or Y 6b is deuterium.
- each Y 1 is the same. In one aspect of these embodiments, each Y 1 is hydrogen. In an alternate aspect of these embodiments, each Y 1 is deuterium. In still another alternate aspect of these embodiments
- each Y 1 is fluoro.
- Y la is hydrogen and Y lb is selected from fluoro and deuterium.
- each Y lb is fluoro.
- Y lb is deuterium.
- each Y 2 is the same. In one aspect of these embodiments, each Y 2 is hydrogen. In an alternate aspect of these embodiments, each Y 2 is deuterium.
- each Y 3 is the same. In one aspect of these embodiments, each Y 3 is hydrogen. In an alternate aspect of these embodiments, each Y 3 is deuterium.
- each Y 4 is the same. In one aspect of these embodiments, each Y 4 is hydrogen. In an alternate aspect of these embodiments, each Y 4 is deuterium.
- each Y 5 is the same. In one aspect of these embodiments, each Y 5 is hydrogen. In an alternate aspect of these embodiments, each Y 5 is deuterium.
- each Y 6 is the same. In one aspect of these embodiments, each Y 6 is hydrogen. In an alternate aspect of these embodiments, each Y 6 is deuterium.
- each Y 1 is the same and is selected from deuterium and fluoro; and each Y 3 and Y 6 is the same (i.e., Y 3a , Y 3b , Y 3c , Y 3d , Y 6a and Y 6b are all the same) and are selected from deuterium and hydrogen.
- each Y 1 is deuterium and each Y 3 and Y 6 is hydrogen.
- each Y 1 is deuterium and each Y 3 and Y 6 is deuterium.
- each Y 1 is the same, each Y 2 is the same, each Y 3 is the same, each Y 4 is the same, each Y 5 is the same, and each Y 6 is the same. In one aspect of these embodiments, each Y 3 and Y 6 is the same. In a more specific aspect of these embodiments, each Y 3 and Y 6 is the same, and each Y 2 and Y 5 is the same.
- the compound of Formula I is not a compound wherein each of Y la , Y lb , Y 2a , Y 2b , Y 3a , Y 3b , Y 3c , Y 3d , Y 4a , Y 4b , Y 5a , Y 5b , Y 6a and Y 6b is deuterium.
- the compound of Formula I is not a compound wherein each of Y la , Y lb is fluorine and each of Y 2a , Y 2b , Y 3a , Y 3b , Y 3c , Y 3d , Y 4a , Y 4b , Y 5a , Y 5b , Y 6a and Y 6b is deuterium.
- each Y 1 is the same, each Y 2 is the same, each Y 3 is the same, each Y 4 is the same, each Y 5 is the same, and each Y 6 is the same and the compound is selected from any one of the compounds (Cmpd) set forth in Table 1 (below):
- any atom not designated as deuterium in any of the embodiments set forth above is present at its natural isotopic abundance.
- Such methods can be carried out utilizing corresponding deuterated and optionally, other isotope-containing reagents and/or intermediates to synthesize the compounds delineated herein, or invoking standard synthetic protocols known in the art for introducing isotopic atoms to a chemical structure.
- the replacement of the reagents set forth in the above patents and patent publication for the corresponding deuterated reagent may be achieved by methods described in one or more of US patent publication Nos. 2006252812, 2011087027, 2011251224, and 2013274483.
- difluorinated benzodioxol reagents such as 5-bromo-2, 2-difluoro-l, 3-benzodioxole, are commercially available and can be used analogously to non-fluorinated reagents by means known in the art of organic synthesis.
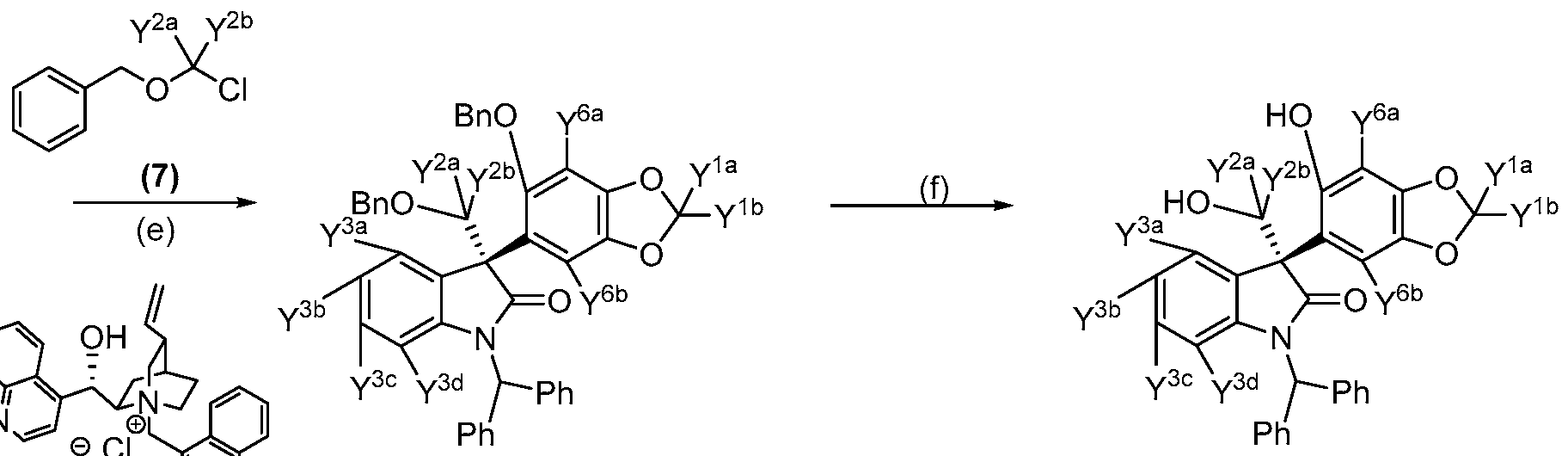
- Reagents and conditions (a) Benzhydryl bromide, CS2CO3; (b) (3), iPr-MgCl; (c) Benzyl bromide, K2CO3; (d) Et 3 SiH, TFA; (e) (7), (8), KOH; (f) H 2 , Pd(OH) 2 /C; (g) DBAD, 2-(Diphenylphosphino) pyridine; (h) TFA, Et 3 SiH; (i) Cs 2 C0 3 .
- appropriately deuterated isatin (1) is alkylated with benzhydryl bromide under basic conditions using a base such as cesium carbonate to afford protected deuterated intermediate (2).
- Condensation with appropriately deuterated 5-Benzodioxolol (3) under Grignard reaction conditions using iPrMgCl in THF provides correspondingly deuterated hydroxyphenol derivative (4).
- Intermediate phenol (4) is treated with benzyl bromide under basic conditions using a base such as K2CO3 at room temperature to provide protected and appropriately deuterated intermediate (5) which is subsequently dehydroxylated with silane reagent, such as triethylsilane, under acidic conditions using acid, such as TFA, to furnish protected and appropriately deuterated intermediate (6).
- silane reagent such as triethylsilane
- deuterated intermediate (1) for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 2.
- isatin intermediate (la) is prepared from commercially available indole-d7 (14a) (98 atom %D) in a manner analogous to the procedure described by Yadav, J. S. et al., Tetrahedron Letters, 48(11), 2029-2032; 2007, by oxidation of (14a) with 2-iodoxybenzoic acid (IBX) in the presence of cerium(III) chloride heptahydrate (CeCh-7H20) at elevated temperature.
- Intermediate (lb) is commercially available or may be similarly prepared from starting material (14b).
- deuterated intermediate (3) for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 3 and Scheme 4.
- Appropriately deuterated intermediate (3a) is prepared from commercially available catechol-de (15a) (98 atom %D) via standard Vilsmeier reaction conditions known in the art, in a manner analogous to the procedure described in Jpn. Kokai Tokkyo Koho, 10001451 to produce intermediate (16a), which is treated with commercially available (17a) (99.5 atom %D) in a manner analogous to the method described in PCT Int. Appl., 2009035652, to furnish correspondingly deuterated intermediate (18a).
- Reagents and conditions (a) 1, 1-thiocarbonylimidazole; (b) 2, 6-dimethyl-tert- butylphenyl sulfur trifluoride, SbCh; (c) TiCU, HF, Br 2 ; (d) KOH, Pd 2 (dba) 3 , tetramethyl di-tBuXPhos
- Intermediate (20a) is brominated at low temperature in a manner analogous to the procedure described in European Patent 1595877 affording intermediate (21a).
- Intermediate (3f) is commercially available or is similarly prepared from commercially available (15b).
- Phase transfer catalyst reagent (8) for use in the preparation of compounds of Formula I according to Scheme 1, is prepared by refluxing a suspension of commercially available starting materials, cinchonine and 9-chloromethylanthracine as described in WO 2013154712 and, by E. J. Corey et al., Organic Synthesis, 2003, 80, 38-45.
- Appropriately deuterated starting materials (24a), (24b) and (24c) may be prepared according to the procedure described in PCT Int. Appl., 2011091035, and then treated with Iridium catalyst at elevated temperature in a manner analogous to the catalytic dehydrogenation procedure described by Yao, W. et al, Angewandte Chemie, International Edition, 53(5), 1390-1394; 2014, to produce furan intermediates (25a), (25b), and (25c).
- transformations and protecting group methodologies useful in synthesizing the applicable compounds are known in the art and include, for example, those described in Larock R, Comprehensive Organic Transformations, VCH Publishers (1989); Greene, TW et al., Protective Groups in Organic Synthesis, 3 rd Ed., John Wiley and Sons (1999); Fieser, L et al., Fieser and Fieser 's Reagents for Organic Synthesis, John Wiley and Sons (1994); and Paquette, L, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995) and subsequent editions thereof.
- the invention also provides pharmaceutical compositions comprising an effective amount of a compound of Formula I (e.g., including any of the formulae herein), or a pharmaceutically acceptable salt of said compound; and a pharmaceutically acceptable carrier.
- a pharmaceutically acceptable carrier e.g., including any of the formulae herein
- the carrier(s) are "acceptable" in the sense of being compatible with the other ingredients of the formulation and, in the case of a pharmaceutically acceptable carrier, not deleterious to the recipient thereof in an amount used in the medicament.
- Pharmaceutically acceptable carriers, adjuvants and vehicles that may be used in the pharmaceutical compositions of this invention include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, di sodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, polyethylene glycol and wool fat.
- ion exchangers alumina, aluminum stearate, lecithin
- serum proteins such as human serum albumin
- buffer substances such as phosphate
- the solubility and bioavailability of the compounds of the present invention in pharmaceutical compositions may be enhanced by methods well-known in the art.
- One method includes the use of lipid excipients in the formulation. See “Oral Lipid-Based Formulations: Enhancing the Bioavailability of Poorly Water-Soluble Drugs (Drugs and the Pharmaceutical Sciences),” David J. Hauss, ed. Informa Healthcare, 2007; and “Role of Lipid Excipients in Modifying Oral and Parenteral Drug Delivery: Basic Principles and Biological Examples," Kishor M. Wasan, ed. Wiley-Interscience, 2006.
- Another known method of enhancing bioavailability is the use of an amorphous form of a compound of this invention optionally formulated with a poloxamer, such as LUTROLTM and PLURONICTM (BASF Corporation), or block copolymers of ethylene oxide and propylene oxide. See United States patent 7,014,866; and United States patent publications 20060094744 and 20060079502.
- compositions of the invention include those suitable for oral, rectal, nasal, topical (including buccal and sublingual), vaginal or parenteral (including subcutaneous, intramuscular, intravenous and intradermal) administration.
- the compound of the formulae herein is administered transdermally (e.g., using a transdermal patch or iontophoretic techniques).
- Other formulations may conveniently be presented in unit dosage form, e.g., tablets, sustained release capsules, and in liposomes, and may be prepared by any methods well known in the art of pharmacy. See, for example, Remington: The Science and Practice of Pharmacy, Lippincott Williams & Wilkins, Baltimore, MD (20th ed. 2000).
- Such preparative methods include the step of bringing into association with the molecule to be administered ingredients such as the carrier that constitutes one or more accessory ingredients.
- ingredients such as the carrier that constitutes one or more accessory ingredients.
- the compositions are prepared by uniformly and intimately bringing into association the active ingredients with liquid carriers, liposomes or finely divided solid carriers, or both, and then, if necessary, shaping the product.
- compositions of the present invention suitable for oral administration may be presented as discrete units such as capsules, sachets, or tablets each containing a predetermined amount of the active ingredient; a powder or granules; a solution or a suspension in an aqueous liquid or a non-aqueous liquid; an oil-in-water liquid emulsion; a water-in-oil liquid emulsion; packed in liposomes; or as a bolus, etc.
- Soft gelatin capsules can be useful for containing such suspensions, which may beneficially increase the rate of compound absorption.
- carriers that are commonly used include lactose and corn starch.
- Lubricating agents such as magnesium stearate, are also typically added.
- useful diluents include lactose and dried cornstarch.
- aqueous suspensions are administered orally, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening and/or flavoring and/or coloring agents may be added.
- compositions suitable for oral administration include lozenges comprising the ingredients in a flavored basis, usually sucrose and acacia or tragacanth; and pastilles comprising the active ingredient in an inert basis such as gelatin and glycerin, or sucrose and acacia.
- compositions suitable for parenteral administration include aqueous and nonaqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the formulations may be presented in unit- dose or multi-dose containers, for example, sealed ampules and vials, and may be stored in a freeze dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
- Such injection solutions may be in the form, for example, of a sterile injectable aqueous or oleaginous suspension.
- This suspension may be formulated according to techniques known in the art using suitable dispersing or wetting agents (such as, for example, Tween 80) and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example, as a solution in 1,3-butanediol.
- the acceptable vehicles and solvents that may be employed are mannitol, water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- Fatty acids, such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions.
- These oil solutions or suspensions may also contain a long- chain alcohol diluent or dispersant.
- compositions of this invention may be administered in the form of suppositories for rectal administration.
- These compositions can be prepared by mixing a compound of this invention with a suitable non-irritating excipient which is solid at room temperature but liquid at the rectal temperature and therefore will melt in the rectum to release the active components.
- suitable non-irritating excipient include, but are not limited to, cocoa butter, beeswax and polyethylene glycols.
- compositions of this invention may be administered by nasal aerosol or inhalation.
- Such compositions are prepared according to techniques well- known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art. See, e.g.: Rabinowitz JD and Zaffaroni AC, US Patent 6,803,031, assigned to Alexza Molecular Delivery Corporation.
- Topical administration of the pharmaceutical compositions of this invention is especially useful when the desired treatment involves areas or organs readily accessible by topical application.
- the pharmaceutical composition should be formulated with a suitable ointment containing the active components suspended or dissolved in a carrier.
- Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petroleum, white petroleum, propylene glycol, polyoxyethylene polyoxypropylene compound, emulsifying wax, and water.
- the pharmaceutical composition can be formulated with a suitable lotion or cream containing the active compound suspended or dissolved in a carrier.
- Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2- octyldodecanol, benzyl alcohol, and water.
- the pharmaceutical compositions of this invention may also be topically applied to the lower intestinal tract by rectal suppository formulation or in a suitable enema formulation. Topically-transdermal patches and iontophoretic administration are also included in this invention.
- Application of the subject therapeutics may be local, so as to be administered at the site of interest.
- Various techniques can be used for providing the subject
- compositions at the site of interest such as injection, use of catheters, trocars, projectiles, pluronic gel, stents, sustained drug release polymers or other device which provides for internal access.
- the compound of the invention is formulated for oral delivery. In other embodiments, the compound of the invention is formulated for topical delivery.
- the invention provides a method of impregnating an implantable drug release device comprising the step of contacting said drug release device with a compound or composition of this invention.
- Implantable drug release devices include, but are not limited to, biodegradable polymer capsules or bullets, non-degradable, diffusible polymer capsules and biodegradable polymer wafers.
- the invention provides an implantable drug release device impregnated with or containing a compound or a composition comprising a compound of this invention, such that said compound is released from said device and is therapeutically active.
- composition of this invention further comprises a second therapeutic agent.
- the second therapeutic agent may be selected from another pain-reducing agent and an agent that is known to have or that demonstrates
- Such agents include steroids, NSAIDs, acetaminophen, other sodium channel blockers known to reduce pain (e.g., procaine, lidocaine, mexiletine), other anti-inflammatory agents, disease-modifying antirheumatic drugs (DMARDs), capsaicin, anticonvulsants (e.g., gabapentin, pregabalin), antidepressants (e.g., nortriptyline, amitriptyline, duloxetine, venlafaxine), and opioid painkillers.
- steroids e.g., NSAIDs, acetaminophen, other sodium channel blockers known to reduce pain (e.g., procaine, lidocaine, mexiletine), other anti-inflammatory agents, disease-modifying antirheumatic drugs (DMARDs), capsaicin, anticonvulsants (e.g., gabapentin, pregabalin), antidepressants (e.g., nortriptyline, amitriptyline
- the invention provides separate dosage forms of a compound of this invention and one or more of any of the above-described second therapeutic agents, wherein the compound and second therapeutic agent are associated with one another.
- association with one another means that the separate dosage forms are packaged together or otherwise attached to one another such that it is readily apparent that the separate dosage forms are intended to be sold and administered together (within less than 24 hours of one another, consecutively or simultaneously).
- the compound of the present invention is present in an effective amount.
- effective amount refers to an amount which, when administered in a proper dosing regimen, is sufficient to treat the target disorder.
- an effective amount of a compound of this invention for topical administration can range from once to twice daily administration of a formulation comprising 0.1% to 10% (w/w) of a compound of the invention. In one aspect of these embodiments, an effective amount of a compound of this invention for topical administration can range from once to twice daily administration of a formulation comprising 0.5% to 8% (w/w) of a compound of the invention.
- Effective doses will also vary, as recognized by those skilled in the art, depending on the diseases treated, the severity of the disease, the route of administration, the sex, age and general health condition of the subject, excipient usage, the possibility of co- usage with other therapeutic treatments such as use of other agents and the judgment of the treating physician.
- an effective amount of the second therapeutic agent is between about 20% and 100% of the dosage normally utilized in a monotherapy regime using just that agent.
- an effective amount is between about 70% and 100% of the normal monotherapeutic dose.
- the normal monotherapeutic dosages of these second therapeutic agents are well known in the art. See, e.g., Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition,
- the invention provides a method of modulating a NaV 1.7 sodium channel in a cell, comprising contacting a cell with one or more compounds of Formula I herein, or a pharmaceutically acceptable salt thereof.
- the method of modulating a NaV 1.7 sodium channel is a method of blocking the channel.
- the invention provides a method of treating a disease or condition that is ameliorated upon blocking a sodium channel by administering to a subject in need thereof a compound of the present invention.
- diseases include, but are not limited to, pain; central nervous conditions such as epilepsy, anxiety, depression and bipolar disease; cardiovascular conditions such as arrhythmias, atrial fibrillation and ventricular fibrillation; neuromuscular conditions such as restless leg syndrome and muscle paralysis or tetanus; stroke, neural trauma, and multiple sclerosis (e.g., to provide neuroprotection against the disease or condition); and channelopathies such as erythromelalgia and familial rectal pain syndrome.
- pain refers to all categories of pain and is recognized to include, but is not limited to, osteoarthritis, dental pain, erythromelalgia, postherpetic neuralgia, neuropathic pain, inflammatory pain, nociceptive pain, idiopathic pain, neuralgic pain, orofacial pain, burn pain, burning mouth syndrome, somatic pain, visceral pain, myofascial pain, dental pain, cancer pain, chemotherapy pain, trauma pain, surgical pain, post-surgical pain, childbirth pain, labor pain, reflex sympathetic dystrophy, brachial plexus avulsion, neurogenic bladder, acute pain (e.g.
- musculoskeletal and post-operative pain chronic pain, persistent pain, peripherally mediated pain, centrally mediated pain, chronic headache, migraine headache, familial hemiplegic migraine, conditions associated with cephalic pain, sinus headache, tension headache, phantom limb pain, peripheral nerve injury, pain following stroke, thalamic lesions, radiculopathy, HIV pain, post-herpetic pain, non-cardiac chest pain, irritable bowel syndrome and pain associated with bowel disorders and dyspepsia, and combinations thereof.
- the disease or condition to be treated is pain.
- the subject is suffering from osteoarthritis, dental pain, erythromelalgia, postherpetic neuralgia and neuropathic pain.
- Identifying a subject in need of such treatment can be in the judgment of a subject or a health care professional and can be subjective (e.g. opinion) or objective (e.g.
- any of the above methods of treatment comprises the further step of co-administering to the subject in need thereof one or more second therapeutic agents.
- the choice of second therapeutic agent may be made from any second therapeutic agent set forth above for use in combination compositions comprising a compound of this invention and a second therapeutic agent.
- co-administered means that the second therapeutic agent may be administered together with a compound of this invention as part of a single dosage form (such as a composition of this invention comprising a compound of the invention and an second therapeutic agent as described above) or as separate, multiple dosage forms. Alternatively, the additional agent may be administered prior to, consecutively with, or following the administration of a compound of this invention. In such combination therapy treatment, both the compounds of this invention and the second therapeutic agent(s) are administered by conventional methods.
- composition of this invention comprising both a compound of the invention and a second therapeutic agent, to a subject does not preclude the separate administration of that same therapeutic agent, any other second therapeutic agent or any compound of this invention to said subject at another time during a course of treatment.
- the effective amount of the compound of this invention is less than its effective amount would be where the second therapeutic agent is not
- the effective amount of the second therapeutic agent is less than its effective amount would be where the compound of this invention is not administered. In this way, undesired side effects associated with high doses of either agent may be minimized.
- Other potential advantages including without limitation improved dosing regimens and/or reduced drug cost) will be apparent to those of skill in the art.
- the invention provides the use of a compound of Formula I alone or together with one or more of the above-described second therapeutic agents in the manufacture of a medicament, either as a single composition or as separate dosage forms, for treatment in a subject of a disease, disorder or symptom set forth above.
- Another aspect of the invention is a compound of Formula I for use in the treatment in a subject of a disease, disorder or symptom thereof delineated herein.
- Microsomal Assay Human liver microsomes (20 mg/mL) are obtained from Xenotech, LLC (Lenexa, KS). ⁇ -nicotinamide adenine dinucleotide phosphate, reduced form (NADPH), magnesium chloride (MgCh), and dimethyl sulfoxide (DMSO) are purchased from Sigma-Aldrich.
- 7.5 mM stock solutions of test compounds are prepared in DMSO.
- the 7.5 mM stock solutions are diluted to 12.5-50 ⁇ in acetonitrile (ACN).
- ACN acetonitrile
- the 20 mg/mL human liver microsomes are diluted to 0.625 mg/mL in 0.1 M potassium phosphate buffer, pH 7.4, containing 3 mM MgCh.
- the diluted microsomes are added to wells of a 96-well deep-well polypropylene plate in triplicate.
- a 10 ⁇ L aliquot of the 12.5-50 ⁇ test compound is added to the microsomes and the mixture is pre-warmed for 10 minutes. Reactions are initiated by addition of pre-warmed NADPH solution.
- the final reaction volume is 0.5 mL and contains 0.5 mg/mL human liver microsomes, 0.25-1.0 ⁇ test compound, and 2 mM NADPH in 0.1 M potassium phosphate buffer, pH 7.4, and 3 mM MgCh.
- the reaction mixtures are incubated at 37 °C, and 50 ⁇ L aliquots are removed at 0, 5, 10, 20, and 30 minutes and added to shallow-well 96-well plates which contain 50 ⁇ L of ice-cold ACN with internal standard to stop the reactions.
- the plates are stored at 4 °C for 20 minutes after which 100 ⁇ L of water is added to the wells of the plate before centrifugation to pellet precipitated proteins.
Abstract
This invention relates to novel, deuterated spiro-oxindole compounds of Formula I, and pharmaceutically acceptable salts thereof. This invention also provides compositions comprising a compound of this invention and the use of such compositions in methods of reducing or eliminating pain and in treating diseases and conditions that are characterized by pain.
Description
DEUTERATED FUNAPIDE AND DIFL U OROFUNAPIDE CROSS REFERENCE TO RELATED APPLICATIONS
[1] This application claims the benefit of U.S. Provisional Serial No. 62/098,670 filed December 31, 2014. This disclosure of the prior application is considered part of (and is incorporated by reference in) the disclosure of this application.
BACKGROUND OF THE INVENTION
[2] Many current medicines suffer from poor absorption, distribution, metabolism and/or excretion (ADME) properties that prevent their wider use or limit their use in certain indications. Poor ADME properties are also a major reason for the failure of drug candidates in clinical trials. While formulation technologies and prodrug strategies can be employed in some cases to improve certain ADME properties, these approaches often fail to address the underlying ADME problems that exist for many drugs and drug candidates. One such problem is rapid metabolism that causes a number of drugs, which otherwise would be highly effective in treating a disease, to be cleared too rapidly from the body. A possible solution to rapid drug clearance is frequent or high dosing to attain a sufficiently high plasma level of drug. This, however, introduces a number of potential treatment problems such as poor patient compliance with the dosing regimen, side effects that become more acute with higher doses, and increased cost of treatment. A rapidly metabolized drug may also expose patients to undesirable toxic or reactive metabolites.
[3] Another ADME limitation that affects many medicines is the formation of toxic or biologically reactive metabolites. As a result, some patients receiving the drug may experience toxicities, or the safe dosing of such drugs may be limited such that patients receive a suboptimal amount of the active agent. In certain cases, modifying dosing intervals or formulation approaches can help to reduce clinical adverse effects, but often the formation of such undesirable metabolites is intrinsic to the metabolism of the compound.
[4] In some select cases, a metabolic inhibitor will be co-administered with a drug that is cleared too rapidly. Such is the case with the protease inhibitor class of drugs that are used to treat HIV infection. The FDA recommends that these drugs be co-dosed with ritonavir, an inhibitor of cytochrome P450 enzyme 3 A4 (CYP3 A4), the enzyme typically
responsible for their metabolism (see Kempf, D.J. et al., Antimicrobial agents and chemotherapy, 1997, 41(3): 654-60). Ritonavir, however, causes adverse effects and adds to the pill burden for HIV patients who must already take a combination of different drugs. Similarly, the CYP2D6 inhibitor quinidine has been added to dextromethorphan for the purpose of reducing rapid CYP2D6 metabolism of dextromethorphan in a treatment of pseudobulbar affect. Quinidine, however, has unwanted side effects that greatly limit its use in potential combination therapy (see Wang, L et al., Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt 1): 659-67; and FDA label for quinidine at www.accessdata.fda.gov).
[5] In general, combining drugs with cytochrome P450 inhibitors is not a satisfactory strategy for decreasing drug clearance. The inhibition of a CYP enzyme's activity can affect the metabolism and clearance of other drugs metabolized by that same enzyme. CYP inhibition can cause other drugs to accumulate in the body to toxic levels.
[6] A potentially attractive strategy for improving a drug's metabolic properties is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the formation of undesirable metabolites by replacing one or more hydrogen atoms with deuterium atoms. Deuterium is a safe, stable, nonradioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the ADME properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
[7] Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, MI et al, J Pharm Sci, 1975, 64:367-91; Foster, AB, Adv Drug Res 1985, 14: 1-40 ("Foster"); Kushner, DJ et al, Can J Physiol Pharmacol 1999, 79-88; Fisher, MB et al, Curr Opin Drug Discov Devel, 2006, 9: 101-09 ("Fisher")). The results have been variable and unpredictable. For some compounds deuteration caused decreased metabolic clearance in vivo. For others, there was no change in metabolism. Still others
demonstrated increased metabolic clearance. The variability in deuterium effects has also led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting adverse metabolism (see Foster at p. 35 and Fisher at p. 101).
[8] The effects of deuterium modification on a drug's metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem. 1991, 34, 2871-76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
SUMMARY OF THE INVENTION
[9] This invention relates to novel, deuterated spiro-oxindole compounds, and pharmaceutically acceptable salts thereof. This invention also provides compositions comprising a compound of this invention and the use of such compositions in methods of reducing or eliminating pain and in treating diseases and conditions that are characterized by pain.
[10] Funapide, also known as TV-45070, XEN-402, and (75)-Γ-[5- (Trifluoromethyl)furan-2-ylmethyl]spiro[furo[2, 3- J[l,3]benzodioxole-7,3'-indol]- 2'(17J)-one is a voltage gated NaV 1.7 sodium channel blocker.
[11] Funapide is currently in Phase II human clinical trials for postherpetic neuralgia, neuropathic pain, osteoarthritic pain, dental pain and erythromelalgia.
[12] Despite the beneficial activities of funapide, there is a continuing need for new compounds to treat the aforementioned diseases and conditions.
DETAILED DESCRIPTION OF THE INVENTION
Definitions
[13] The term "treat" means decrease, suppress, attenuate, diminish, arrest, or stabilize the development or progression of a disease (e.g., a disease or disorder delineated herein), lessen the severity of the disease or improve the symptoms associated with the disease.
[14] "Disease" means any condition or disorder that damages or interferes with the normal function of a cell, tissue, or organ.
[15] It will be recognized that some variation of natural isotopic abundance occurs in a synthesized compound depending upon the origin of chemical materials used in the synthesis. Thus, a preparation of funapide will inherently contain small amounts of deuterated isotopologues. The concentration of naturally abundant stable hydrogen and carbon isotopes, notwithstanding this variation, is small and immaterial as compared to the degree of stable isotopic substitution of compounds of this invention. See, for instance, Wada, E et al., Seikagaku, 1994, 66: 15; Gannes, LZ et al., Comp Biochem Physiol Mol Integr Physiol, 1998, 119:725.
[16] In the compounds of this invention any atom not specifically designated as a particular isotope is meant to represent any stable isotope of that atom. Unless otherwise stated, when a position is designated specifically as "H" or "hydrogen", the position is understood to have hydrogen at its natural abundance isotopic composition. Also unless otherwise stated, when a position is designated specifically as "D" or "deuterium", the position is understood to have deuterium at an abundance that is at least 3340 times greater than the natural abundance of deuterium, which is 0.015% (i.e., at least 50.1% incorporation of deuterium).
[17] The term "isotopic enrichment factor" as used herein means the ratio between the isotopic abundance and the natural abundance of a specified isotope.
[18] In other embodiments, a compound of this invention has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium
incorporation at each designated deuterium atom), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5%) deuterium incorporation).
[19] The term "isotopologue" refers to a species in which the chemical structure differs from a specific compound of this invention only in the isotopic composition thereof.
[20] The term "compound," when referring to a compound of this invention, refers to a collection of molecules having an identical chemical structure, except that there may be isotopic variation among the constituent atoms of the molecules. Thus, it will be clear to those of skill in the art that a compound represented by a particular chemical structure containing indicated deuterium atoms, will also contain lesser amounts of isotopologues having hydrogen atoms at one or more of the designated deuterium positions in that structure. The relative amount of such isotopologues in a compound of this invention will depend upon a number of factors including the isotopic purity of deuterated reagents used to make the compound and the efficiency of incorporation of deuterium in the various synthesis steps used to prepare the compound. However, as set forth above the relative amount of such isotopologues in toto will be less than 49.9% of the compound. In other embodiments, the relative amount of such isotopologues in toto will be less than 47.5%, less than 40%, less than 32.5%, less than 25%, less than 17.5%, less than 10%, less than 5%, less than 3%, less than 1%, or less than 0.5% of the compound.
[21] The invention also provides salts of the compounds of the invention.
[22] A salt of a compound of this invention is formed between an acid and a basic group of the compound, such as an amino functional group, or a base and an acidic group of the compound, such as a carboxyl functional group. According to another
embodiment, the compound is a pharmaceutically acceptable acid addition salt.
[23] The term "pharmaceutically acceptable," as used herein, refers to a component that is, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and other mammals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio. A
"pharmaceutically acceptable salt" means any non-toxic salt that, upon administration to a recipient, is capable of providing, either directly or indirectly, a compound of this invention. A "pharmaceutically acceptable counterion" is an ionic portion of a salt that is not toxic when released from the salt upon administration to a recipient.
[24] Acids commonly employed to form pharmaceutically acceptable salts include inorganic acids such as hydrogen bisulfide, hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid and phosphoric acid, as well as organic acids such as para- toluenesulfonic acid, salicylic acid, tartaric acid, bitartaric acid, ascorbic acid, maleic
acid, besylic acid, fumaric acid, gluconic acid, glucuronic acid, formic acid, glutamic acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, lactic acid, oxalic acid, para-bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid and acetic acid, as well as related inorganic and organic acids. Such
pharmaceutically acceptable salts thus include sulfate, pyrosulfate, bisulfate, sulfite, bisulfite, phosphate, monohydrogenphosphate, dihydrogenphosphate, metaphosphate, pyrophosphate, chloride, bromide, iodide, acetate, propionate, decanoate, caprylate, acrylate, formate, isobutyrate, caprate, heptanoate, propiolate, oxalate, malonate, succinate, suberate, sebacate, fumarate, maleate, butyne-l,4-dioate, hexyne-l,6-dioate, benzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, phthalate, terephthalate, sulfonate, xylene sulfonate, phenyl acetate, phenylpropionate, phenylbutyrate, citrate, lactate, β-hydroxybutyrate, glycolate, maleate, tartrate, methanesulfonate, propanesulfonate, naphthalene- 1 -sulfonate, naphthalene-2- sulfonate, mandelate and other salts. In one embodiment, pharmaceutically acceptable acid addition salts include those formed with mineral acids such as hydrochloric acid and hydrobromic acid, and especially those formed with organic acids such as maleic acid.
[25] The compounds of the present invention (e.g., compounds of Formula I), may contain an asymmetric carbon atom, for example, as the result of deuterium substitution or otherwise. As indicated herein, such stereocenters may be indicated by an asterisk ("*"). As such, compounds of this invention can exist as either individual enantiomers, or mixtures of the two enantiomers. Accordingly, a compound of the present invention may exist as either a racemic mixture or a scalemic mixture, or as individual respective stereoisomers that are substantially free from another possible stereoisomer. The term "substantially free of other stereoisomers" as used herein means less than 25% of other stereoisomers, preferably less than 10% of other stereoisomers, more preferably less than 5% of other stereoisomers and most preferably less than 2% of other stereoisomers are present. Methods of obtaining or synthesizing an individual enantiomer for a given compound are known in the art and may be applied as practicable to final compounds or to starting material or intermediates.
[26] Unless otherwise indicated, when a disclosed compound is named or depicted by a structure without specifying the stereochemistry and has one or more chiral centers, it is
understood to represent all possible stereoisomers of the compound.
[27] The term "stable compounds," as used herein, refers to compounds which possess stability sufficient to allow for their manufacture and which maintain the integrity of the compound for a sufficient period of time to be useful for the purposes detailed herein (e.g., formulation into therapeutic products, intermediates for use in production of therapeutic compounds, isolatable or storable intermediate compounds, treating a disease or condition responsive to therapeutic agents).
[28] "D" and "d" both refer to deuterium. "Stereoisomer" refers to both enantiomers and diastereomers. "Tert" and "t-" each refer to tertiary. "US" refers to the United States of America.
[29] "Substituted with deuterium" refers to the replacement of one or more hydrogen atoms with a corresponding number of deuterium atoms.
[30] Throughout this specification, a variable may be referred to generally (e.g., "each Y3") or may be referred to specifically (e.g., Y3a, Y3b, Y3c, etc.). Unless otherwise indicated, when a variable is referred to generally, it is meant to include all specific embodiments of that particular variable.
Therapeutic Compounds
[31] The present invention provides a compound of Formula I:

each of Yla and Ylb is independently selected from hydrogen, deuterium and fluorine:
each Y2, Y3, Y4, Y5 and Y6 is independently selected from hydrogen and deuterium;
the "*" represents a stereocenter; and
at least one of Yla, Ylb, Y2a, Y2b, Y3a, Y3b, Y3c, Y3d, Y4a, Y4b, Y5a, Y5b, Y6a, or Y6b is deuterium.
[32] In some embodiments of a compound of Formula I, each Y1 is the same. In one aspect of these embodiments, each Y1 is hydrogen. In an alternate aspect of these embodiments, each Y1 is deuterium. In still another alternate aspect of these
embodiments, each Y1 is fluoro.
[33] In other embodiments of a compound of Formula I, Yla is hydrogen and Ylb is selected from fluoro and deuterium. In one aspect of these embodiments, each Ylb is fluoro. In an alternate aspect of these embodiments, Ylb is deuterium.
[34] In some embodiments of a compound of Formula I, each Y2 is the same. In one aspect of these embodiments, each Y2 is hydrogen. In an alternate aspect of these embodiments, each Y2 is deuterium.
[35] In some embodiments of a compound of Formula I, each Y3 is the same. In one aspect of these embodiments, each Y3 is hydrogen. In an alternate aspect of these embodiments, each Y3 is deuterium.
[36] In some embodiments of a compound of Formula I, each Y4 is the same. In one aspect of these embodiments, each Y4 is hydrogen. In an alternate aspect of these embodiments, each Y4 is deuterium.
[37] In some embodiments of a compound of Formula I, each Y5 is the same. In one aspect of these embodiments, each Y5 is hydrogen. In an alternate aspect of these embodiments, each Y5 is deuterium.
[38] In some embodiments of a compound of Formula I, each Y6 is the same. In one aspect of these embodiments, each Y6 is hydrogen. In an alternate aspect of these embodiments, each Y6 is deuterium.
[39] In some embodiments of a compound of Formula I, each Y1 is the same and is selected from deuterium and fluoro; and each Y3 and Y6 is the same (i.e., Y3a, Y3b, Y3c, Y3d, Y6a and Y6b are all the same) and are selected from deuterium and hydrogen. In one aspect of these embodiments, each Y1 is deuterium and each Y3 and Y6 is hydrogen. In
an alternate aspect of these embodiments, each Y1 is deuterium and each Y3 and Y6 is deuterium.
[40] In some embodiments, each Y1 is the same, each Y2 is the same, each Y3 is the same, each Y4 is the same, each Y5 is the same, and each Y6 is the same. In one aspect of these embodiments, each Y3 and Y6 is the same. In a more specific aspect of these embodiments, each Y3 and Y6 is the same, and each Y2 and Y5 is the same.
[41] In one embodiment, the compound of Formula I is not a compound wherein each of Yla, Ylb, Y2a, Y2b, Y3a, Y3b, Y3c, Y3d, Y4a, Y4b, Y5a, Y5b, Y6a and Y6b is deuterium.
[42] In one embodiment, the compound of Formula I is not a compound wherein each of Yla, Ylb is fluorine and each of Y2a, Y2b, Y3a, Y3b, Y3c, Y3d, Y4a, Y4b, Y5a, Y5b, Y6a and Y6b is deuterium.
[43] In one embodiment, each Y1 is the same, each Y2 is the same, each Y3 is the same, each Y4 is the same, each Y5 is the same, and each Y6 is the same and the compound is selected from any one of the compounds (Cmpd) set forth in Table 1 (below):
Table 1 : Exemplary Embodiments of Formula I


[44] In one aspect of each of the above embodiments and aspects thereof, the carbon atom indicated by the "*" is in the (S) configuration and the compound is substantially free of other stereoisomers.
[45] In one embodiment, the following intermediates are also within the scope of this invention.

(6e)
(6f)
[46] In another set of embodiments, any atom not designated as deuterium in any of the embodiments set forth above is present at its natural isotopic abundance.
[47] The synthesis of compounds of Formula I may be readily achieved by synthetic chemists of ordinary skill by reference to the Exemplary Synthesis and Examples disclosed herein. Relevant procedures analogous to those of use for the preparation of compounds of Formula I and intermediates thereof are disclosed, for instance in United
States patent No. US 7,700,641, United States patent publication Nos. 20060252812, and in PCT patent publication Nos. WO 2011047174, WO 2013154712, and WO
2011002708.
[48] Such methods can be carried out utilizing corresponding deuterated and optionally, other isotope-containing reagents and/or intermediates to synthesize the compounds delineated herein, or invoking standard synthetic protocols known in the art for introducing isotopic atoms to a chemical structure. For example, the replacement of the reagents set forth in the above patents and patent publication for the corresponding deuterated reagent may be achieved by methods described in one or more of US patent publication Nos. 2006252812, 2011087027, 2011251224, and 2013274483. In addition, difluorinated benzodioxol reagents, such as 5-bromo-2, 2-difluoro-l, 3-benzodioxole, are commercially available and can be used analogously to non-fluorinated reagents by means known in the art of organic synthesis.
Exemplary Synthesis
[49] A convenient method for synthesizing compounds of Formula I is depicted in Scheme 1 below.
[50] Scheme 1 : General Synthesis of Compounds of Formula I

(5) (6)


Formula I
Reagents and conditions: (a) Benzhydryl bromide, CS2CO3; (b) (3), iPr-MgCl; (c) Benzyl bromide, K2CO3; (d) Et3SiH, TFA; (e) (7), (8), KOH; (f) H2, Pd(OH)2/C; (g) DBAD, 2-(Diphenylphosphino) pyridine; (h) TFA, Et3SiH; (i) Cs2C03.
[51] In a manner analogous to the procedure described in WO2013154712,
appropriately deuterated isatin (1) is alkylated with benzhydryl bromide under basic conditions using a base such as cesium carbonate to afford protected deuterated intermediate (2). Condensation with appropriately deuterated 5-Benzodioxolol (3) under Grignard reaction conditions using iPrMgCl in THF provides correspondingly deuterated
hydroxyphenol derivative (4). Intermediate phenol (4) is treated with benzyl bromide under basic conditions using a base such as K2CO3 at room temperature to provide protected and appropriately deuterated intermediate (5) which is subsequently dehydroxylated with silane reagent, such as triethylsilane, under acidic conditions using acid, such as TFA, to furnish protected and appropriately deuterated intermediate (6). Asymmetric C-alkylation of intermediate (6) using appropriately deuterated benzyl chloromethyl ether (7) and a base, e.g., KOH, under phase transfer conditions in the presence of (,S)-cinchonan-l-ium-9-ol chloride (8) affords fully protected and correspondingly deuterated (^-enantiomer of (benzyloxy)methyl intermediate (9). Intermediate (9) is subjected to hydrogenolysis using Pearlman's catalyst to afford appropriately deuterated dihydroxide (^-enantiomer (10). Intramolecular cylization under Mitsunobu reaction conditions using an azo reagent, e.g., di-tert-butyl
azodicarboxylate, and a phosphine reagent, e.g., 2-(diphenylphosphino)pyridine, at low temperature provides (^-enantiomer, spiro[furo] intermediate (11), which is treated with an acid, e.g., TFA, and a silane reagent, e.g., triethylsilane, to afford the (^-enantiomer of indolone intermediate (12). Subsequent alkylation of intermediate (12) with appropriately deuterated alkyl halide, such as (13), in the presence of a base, e.g., CS2CO3, at elevated temperature produces correspondingly deuterated compounds of Formula I.
[52] It will be appreciated by one skilled in the art that the order of reaction steps may be reversed using synthetic protocols that successfully accomplishes the synthesis of compounds of Formula I.
[53] Compounds of Formula I contain an asymmetric center and a suitable stereoselective procedure for preparation of said compounds is shown in Scheme 1. Compounds of Formula I may also be produced by alternative procedures disclosed for instance in WO 2006110917 and US 2011008727 thereby affording a racemic mixture. When compounds described herein are a racemic mixture, conventional methods such as chiral HPLC or simulated moving bed (SMB) chromatography may be used to resolve the mixture to produce compounds of Formula I.
[54] Using commercially available reagents and deuterated reagents that can be readily prepared by known methods, compounds of Formula I can be prepared with greater than 90% or greater than 95% deuterium incorporation at each position designated as D.
[55] Appropriately deuterated intermediate (1), for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 2.
[56] Scheme 2: Preparation of Intermediate (1)

(1a): each Y3 is D
(14a): each Y3 is D
(1 b): each Y3 is H
(14b): each Y3 is H
Reagents and conditions: CeCh-7H20/IBX
[57] Appropriately deuterated isatin intermediate (la) is prepared from commercially available indole-d7 (14a) (98 atom %D) in a manner analogous to the procedure described by Yadav, J. S. et al., Tetrahedron Letters, 48(11), 2029-2032; 2007, by oxidation of (14a) with 2-iodoxybenzoic acid (IBX) in the presence of cerium(III) chloride heptahydrate (CeCh-7H20) at elevated temperature. Intermediate (lb) is commercially available or may be similarly prepared from starting material (14b).
[58] Appropriately deuterated intermediate (3), for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 3 and Scheme 4.
[59] Scheme 3 : Pre aration of Intermediates 3a, 3b, 3c, 3d)

b . H
 b = D
b = D
Reagents and conditions: (a) DMF, POCb; (b) (17a, X = D) or (17b, X = H), K2CO3; (c)

[60] Appropriately deuterated intermediate (3a) is prepared from commercially available catechol-de (15a) (98 atom %D) via standard Vilsmeier reaction conditions known in the art, in a manner analogous to the procedure described in Jpn. Kokai Tokkyo Koho, 10001451 to produce intermediate (16a), which is treated with commercially available (17a) (99.5 atom %D) in a manner analogous to the method described in PCT Int. Appl., 2009035652, to furnish correspondingly deuterated intermediate (18a).
Subsequent treatment of intermediate (18a) with H2O2 via acid-catalyzed Dakin oxidation affords appropriately deuterated intermediate (3a), also in a manner analogous to PCT Int. Appl., 2009035652. Intermediate (3b) is commercially available, or is similarly prepared from starting material (15b). Intermediate (3c) and Intermediate (3d) are similarly prepared using appropriate reagents.
[61] Scheme 4: Pre aration of Intermediates (3e, 3f)

(15a): Y6a = Y6 = Y6c = Y6d = D (19a): Y6a = Y6 = Y6c = Y6d = D
(15b): Y6a = Y6 = Y6c = Y6d = H (19b): Y6a = Y6 = Y6c = Y6d = H

Reagents and conditions: (a) 1, 1-thiocarbonylimidazole; (b) 2, 6-dimethyl-tert- butylphenyl sulfur trifluoride, SbCh; (c) TiCU, HF, Br2; (d) KOH, Pd2(dba)3, tetramethyl di-tBuXPhos
[62] Appropriately deuterated intermediate (3e) is prepared from commercially available catechol-d6 (15a) (98 atom %D) by treatment with l,l,thiocarbonylimidazole in a manner analogous to the method described by Sugimoto, H., et al., Journal of Organic Chemistry 1988 53 (10), 2263-2267, to produce appropriately deuterated intermediate (19a) which was fluorinated with 4-tert-butyl-2,6-dimethylphenylsulfur trifluoride to afford intermediate (20a) in a manner analogous to the procedure described by Umemoto, T., et al., Journal of the American Chemical Society 2010 132 (51), 18199-18205.
Intermediate (20a) is brominated at low temperature in a manner analogous to the procedure described in European Patent 1595877 affording intermediate (21a). Palladium catalyzed hydroxylation of aryl bromide intermediate (21a) with KOH in a manner analogous to the procedure described in EP 2500345 Al, furnishes appropriately deuterated intermediate (3e). Intermediate (3f) is commercially available or is similarly prepared from commercially available (15b).
[63] Appropriately deuterated intermediate (7), for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 5.
[64] Scheme 5: Preparation of Intermediate (7)

(22) (23a): Y2a = Y2b = D (7a): Y2a = Y2b = D
(23b): Y2a = Y2b = H (7b): Y2a = Y2b = H
Reagents and conditions: (a) HC1
[65] Appropriately deuterated intermediate (7a) is prepared from commercially available benzyl alcohol (22) and paraformaldehyde-d (99+ atom %D) (23a) in a manner analogous to the method described by Connor, D.S. et al., Org. Synth. 1972, 52, 16. Intermediate (7b) is commercially available, or is similarly prepared from commercially available (23b).
[66] Phase transfer catalyst reagent (8), for use in the preparation of compounds of Formula I according to Scheme 1, is prepared by refluxing a suspension of commercially available starting materials, cinchonine and 9-chloromethylanthracine as described in WO 2013154712 and, by E. J. Corey et al., Organic Synthesis, 2003, 80, 38-45.
[67] -l-(anthracen-9-ylmethyl)cinchinonan-l-ium-9-ol chloride (8)

[68] Appropriately deuterated intermediate (13), for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 6.
[69] Scheme 6: Preparation of Intermediate (13)

(25)
(24a)' Y4A = Y4B = Y4C = Y^A =Y^A
_ y5b _ y5b' _ y5c =y5c'— Q
(24b): Y A = Y B = Y C = D; Y5A =
y5a'— y5b _ y5b' _ y5c— y5c'— |_|
(24c): Y A = Y B = Y C= H; Y5A =
y5a'— y5b _ y5b _ Q. y5c— y5c'— |_|

(13a) y4a — y4b— y5a— y5b— Q
(13b) y4a — y4b— Q. y5a— y5b— |_|
(13c) y4a— y4b— |_| . y5a— y5b— Q
Reagents and conditions: (a) Me3CCH=CH2, NaO*Bu, Iridium catalyst; (b) CF3I, TMEDA, Ru(bpy)3Cl2-6H20; (c) N-Bromosuccinimide, AIBN
[70] Appropriately deuterated starting materials (24a), (24b) and (24c) may be prepared according to the procedure described in PCT Int. Appl., 2011091035, and then treated with Iridium catalyst at elevated temperature in a manner analogous to the catalytic dehydrogenation procedure described by Yao, W. et al, Angewandte Chemie, International Edition, 53(5), 1390-1394; 2014, to produce furan intermediates (25a), (25b), and (25c). Alternatively, 2-methylfuran-de (98 atom %D) (25a) and 2- methylfuran-methyl-d3 (99 atom %D) (25b) are commercially available, and furan-2,3,4- d3, 5-methyl-, (25c') wherein Y5c = D, may be prepared as described by Zhou, J. et al., Angewandte Chemie, International Edition, 47(31), 5783-5787; 2008. Treatment of (25a), (25b), and (25c) or (25c') with CF3I via visible light photoredox catalysis in a manner analogous to the procedure described by Iqbal, N., et al., Tetrahedron Letters, 53(15), 2005-2008; 2012, provides intermediates (26a), (26b), (26c) which are subsequently treated with N-bromosuccinimide ( BS) under reaction conditions known in the art to produce appropriately deuterated alkyl bromide (13a), (13b), (13c), or as described by Rajasekhar, M. et al., Journal of Applicable Chemistry (Lumami, India),
3(3), 1260-1267, 8; 2014.
[71] The specific approaches and compounds shown above are not intended to be limiting. The chemical structures in the schemes herein depict variables that are hereby defined commensurately with chemical group definitions (moieties, atoms, etc.) of the corresponding position in the compound formulae herein, whether identified by the same variable name (i.e., Y1, Y2, Y3, etc.) or not. The suitability of a chemical group in a compound structure for use in the synthesis of another compound is within
the knowledge of one of ordinary skill in the art.
[72] Additional methods of synthesizing compounds of Formula I and their synthetic precursors, including those within routes not explicitly shown in schemes herein, are within the means of chemists of ordinary skill in the art. Synthetic chemistry
transformations and protecting group methodologies (protection and deprotection) useful in synthesizing the applicable compounds are known in the art and include, for example, those described in Larock R, Comprehensive Organic Transformations, VCH Publishers (1989); Greene, TW et al., Protective Groups in Organic Synthesis, 3rd Ed., John Wiley and Sons (1999); Fieser, L et al., Fieser and Fieser 's Reagents for Organic Synthesis, John Wiley and Sons (1994); and Paquette, L, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995) and subsequent editions thereof.
[73] Combinations of substituents and variables envisioned by this invention are only those that result in the formation of stable compounds.
Compositions
[74] The invention also provides pharmaceutical compositions comprising an effective amount of a compound of Formula I (e.g., including any of the formulae herein), or a pharmaceutically acceptable salt of said compound; and a pharmaceutically acceptable carrier. The carrier(s) are "acceptable" in the sense of being compatible with the other ingredients of the formulation and, in the case of a pharmaceutically acceptable carrier, not deleterious to the recipient thereof in an amount used in the medicament.
[75] Pharmaceutically acceptable carriers, adjuvants and vehicles that may be used in the pharmaceutical compositions of this invention include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum
albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, di sodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, polyethylene glycol and wool fat.
[76] If required, the solubility and bioavailability of the compounds of the present invention in pharmaceutical compositions may be enhanced by methods well-known in the art. One method includes the use of lipid excipients in the formulation. See "Oral Lipid-Based Formulations: Enhancing the Bioavailability of Poorly Water-Soluble Drugs (Drugs and the Pharmaceutical Sciences)," David J. Hauss, ed. Informa Healthcare, 2007; and "Role of Lipid Excipients in Modifying Oral and Parenteral Drug Delivery: Basic Principles and Biological Examples," Kishor M. Wasan, ed. Wiley-Interscience, 2006.
[77] Another known method of enhancing bioavailability is the use of an amorphous form of a compound of this invention optionally formulated with a poloxamer, such as LUTROL™ and PLURONIC™ (BASF Corporation), or block copolymers of ethylene oxide and propylene oxide. See United States patent 7,014,866; and United States patent publications 20060094744 and 20060079502.
[78] The pharmaceutical compositions of the invention include those suitable for oral, rectal, nasal, topical (including buccal and sublingual), vaginal or parenteral (including subcutaneous, intramuscular, intravenous and intradermal) administration. In certain embodiments, the compound of the formulae herein is administered transdermally (e.g., using a transdermal patch or iontophoretic techniques). Other formulations may conveniently be presented in unit dosage form, e.g., tablets, sustained release capsules, and in liposomes, and may be prepared by any methods well known in the art of pharmacy. See, for example, Remington: The Science and Practice of Pharmacy, Lippincott Williams & Wilkins, Baltimore, MD (20th ed. 2000).
[79] Such preparative methods include the step of bringing into association with the molecule to be administered ingredients such as the carrier that constitutes one or more accessory ingredients. In general, the compositions are prepared by uniformly and
intimately bringing into association the active ingredients with liquid carriers, liposomes or finely divided solid carriers, or both, and then, if necessary, shaping the product.
[80] In certain embodiments, the compound is administered orally. Compositions of the present invention suitable for oral administration may be presented as discrete units such as capsules, sachets, or tablets each containing a predetermined amount of the active ingredient; a powder or granules; a solution or a suspension in an aqueous liquid or a non-aqueous liquid; an oil-in-water liquid emulsion; a water-in-oil liquid emulsion; packed in liposomes; or as a bolus, etc. Soft gelatin capsules can be useful for containing such suspensions, which may beneficially increase the rate of compound absorption.
[81] In the case of tablets for oral use, carriers that are commonly used include lactose and corn starch. Lubricating agents, such as magnesium stearate, are also typically added. For oral administration in a capsule form, useful diluents include lactose and dried cornstarch. When aqueous suspensions are administered orally, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening and/or flavoring and/or coloring agents may be added.
[82] Compositions suitable for oral administration include lozenges comprising the ingredients in a flavored basis, usually sucrose and acacia or tragacanth; and pastilles comprising the active ingredient in an inert basis such as gelatin and glycerin, or sucrose and acacia.
[83] Compositions suitable for parenteral administration include aqueous and nonaqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents. The formulations may be presented in unit- dose or multi-dose containers, for example, sealed ampules and vials, and may be stored in a freeze dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
[84] Such injection solutions may be in the form, for example, of a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to
techniques known in the art using suitable dispersing or wetting agents (such as, for example, Tween 80) and suspending agents. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example, as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that may be employed are mannitol, water, Ringer's solution and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose, any bland fixed oil may be employed including synthetic mono- or diglycerides. Fatty acids, such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions. These oil solutions or suspensions may also contain a long- chain alcohol diluent or dispersant.
[85] The pharmaceutical compositions of this invention may be administered in the form of suppositories for rectal administration. These compositions can be prepared by mixing a compound of this invention with a suitable non-irritating excipient which is solid at room temperature but liquid at the rectal temperature and therefore will melt in the rectum to release the active components. Such materials include, but are not limited to, cocoa butter, beeswax and polyethylene glycols.
[86] The pharmaceutical compositions of this invention may be administered by nasal aerosol or inhalation. Such compositions are prepared according to techniques well- known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art. See, e.g.: Rabinowitz JD and Zaffaroni AC, US Patent 6,803,031, assigned to Alexza Molecular Delivery Corporation.
[87] Topical administration of the pharmaceutical compositions of this invention is especially useful when the desired treatment involves areas or organs readily accessible by topical application. For topical application topically to the skin, the pharmaceutical composition should be formulated with a suitable ointment containing the active components suspended or dissolved in a carrier. Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petroleum,
white petroleum, propylene glycol, polyoxyethylene polyoxypropylene compound, emulsifying wax, and water. Alternatively, the pharmaceutical composition can be formulated with a suitable lotion or cream containing the active compound suspended or dissolved in a carrier. Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2- octyldodecanol, benzyl alcohol, and water. The pharmaceutical compositions of this invention may also be topically applied to the lower intestinal tract by rectal suppository formulation or in a suitable enema formulation. Topically-transdermal patches and iontophoretic administration are also included in this invention.
[88] Application of the subject therapeutics may be local, so as to be administered at the site of interest. Various techniques can be used for providing the subject
compositions at the site of interest, such as injection, use of catheters, trocars, projectiles, pluronic gel, stents, sustained drug release polymers or other device which provides for internal access.
[89] In some embodiments, the compound of the invention is formulated for oral delivery. In other embodiments, the compound of the invention is formulated for topical delivery.
[90] According to another embodiment, the invention provides a method of impregnating an implantable drug release device comprising the step of contacting said drug release device with a compound or composition of this invention. Implantable drug release devices include, but are not limited to, biodegradable polymer capsules or bullets, non-degradable, diffusible polymer capsules and biodegradable polymer wafers.
[91] According to another embodiment, the invention provides an implantable drug release device impregnated with or containing a compound or a composition comprising a compound of this invention, such that said compound is released from said device and is therapeutically active.
[92] In another embodiment, a composition of this invention further comprises a second therapeutic agent. The second therapeutic agent may be selected from another pain-reducing agent and an agent that is known to have or that demonstrates
advantageous properties in the treatment of a non-pain aspect of the disease to be treated. Such agents include steroids, NSAIDs, acetaminophen, other sodium channel blockers
known to reduce pain (e.g., procaine, lidocaine, mexiletine), other anti-inflammatory agents, disease-modifying antirheumatic drugs (DMARDs), capsaicin, anticonvulsants (e.g., gabapentin, pregabalin), antidepressants (e.g., nortriptyline, amitriptyline, duloxetine, venlafaxine), and opioid painkillers.
[93] In another embodiment, the invention provides separate dosage forms of a compound of this invention and one or more of any of the above-described second therapeutic agents, wherein the compound and second therapeutic agent are associated with one another. The term "associated with one another" as used herein means that the separate dosage forms are packaged together or otherwise attached to one another such that it is readily apparent that the separate dosage forms are intended to be sold and administered together (within less than 24 hours of one another, consecutively or simultaneously).
[94] In the pharmaceutical compositions of the invention, the compound of the present invention is present in an effective amount. As used herein, the term "effective amount" refers to an amount which, when administered in a proper dosing regimen, is sufficient to treat the target disorder.
[95] The interrelationship of dosages for animals and humans (based on milligrams per meter squared of body surface) is described in Freireich et al., Cancer Chemother. Rep, 1966, 50: 219. Body surface area may be approximately determined from height and weight of the subject. See, e.g., Scientific Tables, Geigy Pharmaceuticals, Ardsley, N.Y., 1970, 537.
[96] In some embodiments, an effective amount of a compound of this invention for topical administration can range from once to twice daily administration of a formulation comprising 0.1% to 10% (w/w) of a compound of the invention. In one aspect of these embodiments, an effective amount of a compound of this invention for topical administration can range from once to twice daily administration of a formulation comprising 0.5% to 8% (w/w) of a compound of the invention.
[97] Effective doses will also vary, as recognized by those skilled in the art, depending on the diseases treated, the severity of the disease, the route of administration, the sex, age and general health condition of the subject, excipient usage, the possibility of co-
usage with other therapeutic treatments such as use of other agents and the judgment of the treating physician.
[98] For pharmaceutical compositions that comprise a second therapeutic agent, an effective amount of the second therapeutic agent is between about 20% and 100% of the dosage normally utilized in a monotherapy regime using just that agent. Preferably, an effective amount is between about 70% and 100% of the normal monotherapeutic dose. The normal monotherapeutic dosages of these second therapeutic agents are well known in the art. See, e.g., Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition,
Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), each of which references are incorporated herein by reference in their entirety.
[99] It is expected that some of the second therapeutic agents referenced above will act synergistically with the compounds of this invention. When this occurs, it will allow the effective dosage of the second therapeutic agent and/or the compound of this invention to be reduced from that required in a monotherapy. This has the advantage of minimizing toxic side effects of either the second therapeutic agent of a compound of this invention, synergistic improvements in efficacy, improved ease of administration or use and/or reduced overall expense of compound preparation or formulation.
Methods of Treatment
[100] In another embodiment, the invention provides a method of modulating a NaV 1.7 sodium channel in a cell, comprising contacting a cell with one or more compounds of Formula I herein, or a pharmaceutically acceptable salt thereof. In some embodiments, the method of modulating a NaV 1.7 sodium channel is a method of blocking the channel.
[101] According to another embodiment, the invention provides a method of treating a disease or condition that is ameliorated upon blocking a sodium channel by administering to a subject in need thereof a compound of the present invention. Such diseases include, but are not limited to, pain; central nervous conditions such as epilepsy, anxiety, depression and bipolar disease; cardiovascular conditions such as arrhythmias, atrial fibrillation and ventricular fibrillation; neuromuscular conditions such as restless leg
syndrome and muscle paralysis or tetanus; stroke, neural trauma, and multiple sclerosis (e.g., to provide neuroprotection against the disease or condition); and channelopathies such as erythromelalgia and familial rectal pain syndrome.
[102] The term "pain" refers to all categories of pain and is recognized to include, but is not limited to, osteoarthritis, dental pain, erythromelalgia, postherpetic neuralgia, neuropathic pain, inflammatory pain, nociceptive pain, idiopathic pain, neuralgic pain, orofacial pain, burn pain, burning mouth syndrome, somatic pain, visceral pain, myofascial pain, dental pain, cancer pain, chemotherapy pain, trauma pain, surgical pain, post-surgical pain, childbirth pain, labor pain, reflex sympathetic dystrophy, brachial plexus avulsion, neurogenic bladder, acute pain (e.g. musculoskeletal and post-operative pain), chronic pain, persistent pain, peripherally mediated pain, centrally mediated pain, chronic headache, migraine headache, familial hemiplegic migraine, conditions associated with cephalic pain, sinus headache, tension headache, phantom limb pain, peripheral nerve injury, pain following stroke, thalamic lesions, radiculopathy, HIV pain, post-herpetic pain, non-cardiac chest pain, irritable bowel syndrome and pain associated with bowel disorders and dyspepsia, and combinations thereof.
[103] In some embodiments, the disease or condition to be treated is pain. In one aspect of these embodiments, the subject is suffering from osteoarthritis, dental pain, erythromelalgia, postherpetic neuralgia and neuropathic pain.
[104] Identifying a subject in need of such treatment can be in the judgment of a subject or a health care professional and can be subjective (e.g. opinion) or objective (e.g.
measurable by a test or diagnostic method).
[105] In another embodiment, any of the above methods of treatment comprises the further step of co-administering to the subject in need thereof one or more second therapeutic agents. The choice of second therapeutic agent may be made from any second therapeutic agent set forth above for use in combination compositions comprising a compound of this invention and a second therapeutic agent.
[106] The term "co-administered" as used herein means that the second therapeutic agent may be administered together with a compound of this invention as part of a single dosage form (such as a composition of this invention comprising a compound of the invention and an second therapeutic agent as described above) or as separate, multiple
dosage forms. Alternatively, the additional agent may be administered prior to, consecutively with, or following the administration of a compound of this invention. In such combination therapy treatment, both the compounds of this invention and the second therapeutic agent(s) are administered by conventional methods. The administration of a composition of this invention, comprising both a compound of the invention and a second therapeutic agent, to a subject does not preclude the separate administration of that same therapeutic agent, any other second therapeutic agent or any compound of this invention to said subject at another time during a course of treatment.
[107] Effective amounts of these second therapeutic agents are well known to those skilled in the art and guidance for dosing may be found in patents and published patent applications referenced herein, as well as in Wells et al., eds., Pharmacotherapy
Handbook, 2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR
Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), and other medical texts. However, it is well within the skilled artisan's purview to determine the second therapeutic agent's optimal effective-amount range.
[108] In one embodiment of the invention, where a second therapeutic agent is administered to a subject, the effective amount of the compound of this invention is less than its effective amount would be where the second therapeutic agent is not
administered. In another embodiment, the effective amount of the second therapeutic agent is less than its effective amount would be where the compound of this invention is not administered. In this way, undesired side effects associated with high doses of either agent may be minimized. Other potential advantages (including without limitation improved dosing regimens and/or reduced drug cost) will be apparent to those of skill in the art.
[109] In yet another aspect, the invention provides the use of a compound of Formula I alone or together with one or more of the above-described second therapeutic agents in the manufacture of a medicament, either as a single composition or as separate dosage forms, for treatment in a subject of a disease, disorder or symptom set forth above.
Another aspect of the invention is a compound of Formula I for use in the treatment in a
subject of a disease, disorder or symptom thereof delineated herein. Example X. Evaluation of Metabolic Stability
[110] Microsomal Assay: Human liver microsomes (20 mg/mL) are obtained from Xenotech, LLC (Lenexa, KS). β-nicotinamide adenine dinucleotide phosphate, reduced form (NADPH), magnesium chloride (MgCh), and dimethyl sulfoxide (DMSO) are purchased from Sigma-Aldrich.
[Ill] Determination of Metabolic Stability: 7.5 mM stock solutions of test compounds are prepared in DMSO. The 7.5 mM stock solutions are diluted to 12.5-50 μΜ in acetonitrile (ACN). The 20 mg/mL human liver microsomes are diluted to 0.625 mg/mL in 0.1 M potassium phosphate buffer, pH 7.4, containing 3 mM MgCh. The diluted microsomes are added to wells of a 96-well deep-well polypropylene plate in triplicate. A 10 μL aliquot of the 12.5-50 μΜ test compound is added to the microsomes and the mixture is pre-warmed for 10 minutes. Reactions are initiated by addition of pre-warmed NADPH solution. The final reaction volume is 0.5 mL and contains 0.5 mg/mL human liver microsomes, 0.25-1.0 μΜ test compound, and 2 mM NADPH in 0.1 M potassium phosphate buffer, pH 7.4, and 3 mM MgCh. The reaction mixtures are incubated at 37 °C, and 50 μL aliquots are removed at 0, 5, 10, 20, and 30 minutes and added to shallow-well 96-well plates which contain 50 μL of ice-cold ACN with internal standard to stop the reactions. The plates are stored at 4 °C for 20 minutes after which 100 μL of water is added to the wells of the plate before centrifugation to pellet precipitated proteins. Supernatants are transferred to another 96-well plate and analyzed for amounts of parent remaining by LC-MS/MS using an Applied Bio-systems API 4000 mass spectrometer. The same procedure is followed for the non-deuterated counterpart of the compound of Formula I and the positive control, 7-ethoxycoumarin (1 μΜ). Testing is done in triplicate.
[112] Data analysis: The in vitro t s for test compounds are calculated from the slopes of the linear regression of % parent remaining (In) vs incubation time relationship,
in vitro t ½ = 0.693/k
k = -[slope of linear regression of % parent remaining (In) vs incubation time]
[113] Data analysis is performed using Microsoft Excel Software.
[114] Without further description, it is believed that one of ordinary skill in the art can, using the preceding description and the illustrative examples, make and utilize the compounds of the present invention and practice the claimed methods. It should be understood that the foregoing discussion and examples merely present a detailed description of certain preferred embodiments. It will be apparent to those of ordinary skill in the art that various modifications and equivalents can be made without departing from the spirit and scope of the invention.
Claims
1. A com ound of Formula I:

each Y1 is independently selected from hydrogen, deuterium and fluoro;
each Y2, Y3, Y4, Y5 and Y6 is independently selected from hydrogen and deuterium;
the "*" represents a stereocenter; and
at least one of Yla, Ylb, Y2a, Y2b, Y3a, Y3b, Y3c, Y3d, Y4a, Y4b, Y5a, Y5b, Y6a, or Y< is deuterium.
2. The compound of claim 1, wherein:
each Y1 is the same;
each Y2 is the same;
each Y3 is the same;
each Y4 is the same;
each Y5 is the same; and
each Y6 is the same.
3. The compound of claim 1 or 2, wherein Y a and Y are the same and are selected from deuterium and fluoro.
4. The compound of any one of claims 1-3, wherein Y , Y , Y , Y , Y and Y' are the same.
5. The compound of claim 2, wherein the compound is selected from any one of the compounds set forth in table below:

or a pharmaceutically acceptable salt thereof, wherein any atom not specifical designated as deuterium is present at its natural isotopic abundance.
6. The compound of any one of claims 1-5, wherein the carbon atom indicated by the "*" is in the (S) configuration and the compound is substantially free of other stereoisomers.
7. The compound of any one of claims 1-6, wherein any atom not designated as deuterium is present at its natural isotopic abundance.
8. A pharmaceutical composition comprising an effective amount of the compound of any one of claims 1-7, or a pharmaceutically acceptable salt thereof; and a
pharmaceutically acceptable carrier.
9. The pharmaceutical composition of claim 8, wherein the compound is formulated for topical administration.
10. The pharmaceutical composition of claim 9, wherein the compound is formulated as an ointment.
11. The pharmaceutical composition of claim 8, wherein the compound is formulated for oral administration.
12. A method of blocking a NaV 1.7 sodium channel in a cell comprising the step of contacting the cell with a compound of any one of claims 1-7.
13. A method of treating a disease or condition selected from pain; central nervous conditions; cardiovascular conditions; neuromuscular conditions; stroke; neural trauma; multiple sclerosis; and channelopathies comprising the step of administering to a subject in need thereof the composition of any one of claims 8-11.
14. The method of claim 13, wherein the condition is selected from osteoarthritis, dental pain, erythromelalgia, postherpetic neuralgia, neuropathic pain, inflammatory pain, nociceptive pain, idiopathic pain, neuralgic pain, orofacial pain, burn pain, burning mouth syndrome, somatic pain, visceral pain, myofascial pain, dental pain, cancer pain, chemotherapy pain, trauma pain, surgical pain, post-surgical pain, childbirth pain, labor pain, reflex sympathetic dystrophy, brachial plexus avulsion, neurogenic bladder, acute pain, chronic pain, persistent pain, peripherally mediated pain, centrally mediated pain,
chronic headache, migraine headache, familial hemiplegic migraine, conditions associated with cephalic pain, sinus headache, tension headache, phantom limb pain, peripheral nerve injury, pain following stroke, thalamic lesions, radiculopathy, HIV pain, post-herpetic pain, non-cardiac chest pain, irritable bowel syndrome and pain associated with bowel disorders and dyspepsia, and combinations thereof.
15. The method of claim 14, wherein the condition is selected from osteoarthritis, dental pain, erythromelalgia, postherpetic neuralgia and neuropathic pain.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462098670P | 2014-12-31 | 2014-12-31 | |
| US62/098,670 | 2014-12-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016109795A1 true WO2016109795A1 (en) | 2016-07-07 |
Family
ID=55221547
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/068276 WO2016109795A1 (en) | 2014-12-31 | 2015-12-31 | Deuterated funapide and difluorofunapide |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2016109795A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114716380A (en) * | 2022-04-01 | 2022-07-08 | 新乡医学院 | Efficient phase transfer catalytic 4-substituted pyrazolone compound asymmetric fluorination method |
| CN116120272A (en) * | 2022-12-31 | 2023-05-16 | 淮北师范大学 | Method for synthesizing fludioxonil intermediate 2, 2-difluoro-1, 3-benzodioxole-4-formaldehyde |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6803031B2 (en) | 2001-05-24 | 2004-10-12 | Alexza Molecular Delivery Corporation | Delivery of erectile dysfunction drugs through an inhalation route |
| EP1595877A1 (en) | 2004-05-14 | 2005-11-16 | Lanxess Deutschland GmbH | Process for the preparation of 5-bromo-2,2-difluorobenzo-(1,3)-dioxoles |
| US7014866B2 (en) | 2001-05-03 | 2006-03-21 | Hoffmann-La Roche Inc. | High dose solid unit oral pharmaceutical dosage form of amorphous nelfinavir mesylate and process for making same |
| US20060079502A1 (en) | 1999-11-02 | 2006-04-13 | Steffen Lang | Pharmaceutical compositions |
| US20060094744A1 (en) | 2004-09-29 | 2006-05-04 | Maryanoff Cynthia A | Pharmaceutical dosage forms of stable amorphous rapamycin like compounds |
| WO2006110917A2 (en) | 2005-04-11 | 2006-10-19 | Xenon Pharmaceuticals Inc. | Spiro-oxindole compounds and their uses as therapeutic agents |
| WO2009035652A1 (en) | 2007-09-13 | 2009-03-19 | Concert Pharmaceuticals, Inc. | Synthesis of deuterated catechols and benzo[d][1,3] dioxoles and derivatives thereof |
| WO2011002708A1 (en) | 2009-06-29 | 2011-01-06 | Xenon Pharmaceuticals Inc. | Enantiomers of spiro-oxindole compounds and their uses as therapeutic agents |
| US20110008727A1 (en) | 2006-05-26 | 2011-01-13 | International Business Machines Corporation | Low Activation Energy Photoresist Composition and Process for Its Use |
| US20110087027A1 (en) | 2009-10-14 | 2011-04-14 | Xenon Pharmaceuticals Inc. | Synthetic methods for spiro-oxindole compounds |
| WO2011091035A1 (en) | 2010-01-19 | 2011-07-28 | Concert Pharmaceuticals, Inc. | Aminoquinoline derivatives |
| EP2500345A1 (en) | 2009-11-11 | 2012-09-19 | Dainippon Sumitomo Pharma Co., Ltd. | 8-azabicyclo[3.2.1]octane-8-carboxamide derivative |
| WO2013154712A1 (en) | 2012-04-12 | 2013-10-17 | Xenon Pharmaceuticals Inc. | Asymmetric syntheses for spiro-oxindole compounds useful as therapeutic agents |
-
2015
- 2015-12-31 WO PCT/US2015/068276 patent/WO2016109795A1/en active Application Filing
Patent Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060079502A1 (en) | 1999-11-02 | 2006-04-13 | Steffen Lang | Pharmaceutical compositions |
| US7014866B2 (en) | 2001-05-03 | 2006-03-21 | Hoffmann-La Roche Inc. | High dose solid unit oral pharmaceutical dosage form of amorphous nelfinavir mesylate and process for making same |
| US6803031B2 (en) | 2001-05-24 | 2004-10-12 | Alexza Molecular Delivery Corporation | Delivery of erectile dysfunction drugs through an inhalation route |
| EP1595877A1 (en) | 2004-05-14 | 2005-11-16 | Lanxess Deutschland GmbH | Process for the preparation of 5-bromo-2,2-difluorobenzo-(1,3)-dioxoles |
| US20060094744A1 (en) | 2004-09-29 | 2006-05-04 | Maryanoff Cynthia A | Pharmaceutical dosage forms of stable amorphous rapamycin like compounds |
| US7700641B2 (en) | 2005-04-11 | 2010-04-20 | Xenon Pharmaceuticals Inc. | Spiro-oxindole compounds and their uses as therapeutic agents |
| US20060252812A1 (en) | 2005-04-11 | 2006-11-09 | Xenon Pharmaceuticals Inc. | Spiro-oxindole compounds and their uses as therapeutic agents |
| WO2006110917A2 (en) | 2005-04-11 | 2006-10-19 | Xenon Pharmaceuticals Inc. | Spiro-oxindole compounds and their uses as therapeutic agents |
| US20110251224A1 (en) | 2005-04-11 | 2011-10-13 | Xenon Pharmaceuticals Inc. | Spiro-oxindole compounds and their uses as therapeutic agents |
| US20110008727A1 (en) | 2006-05-26 | 2011-01-13 | International Business Machines Corporation | Low Activation Energy Photoresist Composition and Process for Its Use |
| WO2009035652A1 (en) | 2007-09-13 | 2009-03-19 | Concert Pharmaceuticals, Inc. | Synthesis of deuterated catechols and benzo[d][1,3] dioxoles and derivatives thereof |
| WO2011002708A1 (en) | 2009-06-29 | 2011-01-06 | Xenon Pharmaceuticals Inc. | Enantiomers of spiro-oxindole compounds and their uses as therapeutic agents |
| US20110087027A1 (en) | 2009-10-14 | 2011-04-14 | Xenon Pharmaceuticals Inc. | Synthetic methods for spiro-oxindole compounds |
| WO2011047174A1 (en) | 2009-10-14 | 2011-04-21 | Xenon Pharmaceuticals Inc. | Synthetic methods for spiro-oxindole compounds |
| EP2500345A1 (en) | 2009-11-11 | 2012-09-19 | Dainippon Sumitomo Pharma Co., Ltd. | 8-azabicyclo[3.2.1]octane-8-carboxamide derivative |
| WO2011091035A1 (en) | 2010-01-19 | 2011-07-28 | Concert Pharmaceuticals, Inc. | Aminoquinoline derivatives |
| WO2013154712A1 (en) | 2012-04-12 | 2013-10-17 | Xenon Pharmaceuticals Inc. | Asymmetric syntheses for spiro-oxindole compounds useful as therapeutic agents |
| US20130274483A1 (en) | 2012-04-12 | 2013-10-17 | Xenon Pharmaceuticals Inc. | Asymmetric syntheses for spiro-oxindole compounds useful as therapeutic agents |
Non-Patent Citations (31)
| Title |
|---|
| "Encyclopedia ofReagents for Organic Synthesis", 1995, JOHN WILEY AND SONS |
| "Geigy Pharmaceuticals", 1970, ARDSLEY, pages: 537 |
| "Oral Lipid-Based Formulations: Enhancing the Bioavailability of Poorly Water-Soluble Drugs (Drugs and the Pharmaceutical Sciences", 2007, INFORMA HEALTHCARE |
| "PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000", 2000, TARASCON PUBLISHING |
| "Pharmacotherapy Handbook", 2000, APPLETON AND LANGE |
| "Remington: The Science and Practice of Pharmacy", 2000, LIPPINCOTT WILLIAMS & WILKINS |
| "Role of Lipid Excipients in Modifying Oral and Parenteral Drug Delivery: Basic Principles and Biological Examples", 2006, WILEY-INTERSCIENCE |
| BLAKE, MI ET AL., J PHARM SCI, vol. 64, 1975, pages 367 - 391 |
| CONNOR, D.S. ET AL., ORG. SYNTH., vol. 52, 1972, pages 16 |
| E. J. COREY ET AL., ORGANIC SYNTHESIS, vol. 80, 2003, pages 38 - 45 |
| FIESER, L ET AL.: "Fieser and Fieser's Reagents for Organic Synthesis", JOHN WILEY AND SONS |
| FISHER, MB ET AL., CURR OPIN DRUG DISCOV DEVEL, vol. 9, 2006, pages 101 - 109 |
| FOSTER A B: "Deuterium isotope effects in the metabolism of drugs and xenobiotics: implications for drug design", ADVANCES IN DRUG RESEARCH, ACADEMIC PRESS, LONDON, GB, vol. 14, 1 January 1985 (1985-01-01), pages 1 - 40, XP009086953, ISSN: 0065-2490 * |
| FOSTER, AB, ADV DRUG RES, vol. 14, 1985, pages 1 - 40 |
| FREIREICH ET AL., CANCER CHEMOTHER. REP, vol. 50, 1966, pages 219 |
| FUKUTO ET AL., J. MED. CHEM., vol. 34, 1991, pages 2871 - 2876 |
| GANNES, LZ ET AL., COMP BIOCHEM PHYSIOL MOL INTEGR PHYSIOL, vol. 119, 1998, pages 725 |
| GREENE, TW ET AL.: "Protective Groups in Organic Synthesis. 3rd ed.", 1999, JOHN WILEY AND SONS |
| IQBAL, N. ET AL., TETRAHEDRON LETTERS, vol. 53, no. 15, 2012, pages 2005 - 2008 |
| JON M. FUKUTO ET AL: "Determination of the mechanism of demethylenation of (methylenedioxy)phenyl compounds by cytochrome P450 using deuterium isotope effects", JOURNAL OF MEDICINAL CHEMISTRY, vol. 34, no. 9, 1 September 1991 (1991-09-01), pages 2871 - 2876, XP055009050, ISSN: 0022-2623, DOI: 10.1021/jm00113a028 * |
| KEMPF, D.J ET AL., ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, vol. 41, no. 3, 1997, pages 654 - 660 |
| KUSHNER, DJ ET AL., CAN J PHYSIOL PHARMACOL, 1999, pages 79 - 88 |
| LAROCK R: "Comprehensive Organic Transformations", 1989, VCH PUBLISHERS |
| RAJASEKHAR, M. ET AL., JOURNAL OF APPLICABLE CHEMISTRY (LUMAMI, INDIA, vol. 3, no. 3, 2014, pages 1260 - 1267 |
| SUGIMOTO, H. ET AL., JOURNAL OF ORGANIC CHEMISTRY, vol. 53, no. 10, 1988, pages 2263 - 2267 |
| UMEMOTO, T. ET AL., JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 132, no. 51, 2010, pages 18199 - 18205 |
| WADA, E ET AL., SEIKAGAKU, vol. 66, 1994, pages 15 |
| WANG, L ET AL., CLINICAL PHARMACOLOGY AND THERAPEUTICS, vol. 56, 1994, pages 659 - 667 |
| YADAV, J. S. ET AL., TETRAHEDRON LETTERS, vol. 48, no. 11, 2007, pages 2029 - 2032 |
| YAO, W ET AL., ANGEWANDTE CHEMIE, INTERNATIONAL EDITION, vol. 53, no. 5, 2014, pages 1390 - 1394 |
| ZHOU, J. ET AL., ANGEWANDTE CHEMIE, INTERNATIONAL EDITION, vol. 47, no. 31, 2008, pages 5783 - 5787 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114716380A (en) * | 2022-04-01 | 2022-07-08 | 新乡医学院 | Efficient phase transfer catalytic 4-substituted pyrazolone compound asymmetric fluorination method |
| CN116120272A (en) * | 2022-12-31 | 2023-05-16 | 淮北师范大学 | Method for synthesizing fludioxonil intermediate 2, 2-difluoro-1, 3-benzodioxole-4-formaldehyde |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8471034B2 (en) | Niacin prodrugs and deuterated versions thereof | |
| JP5852565B2 (en) | Deuterated isoindoline-1,3-dione derivatives as PDE4 and TNF-α inhibitors | |
| WO2012151361A1 (en) | Carbamoylpyridone derivatives | |
| EP3450434A1 (en) | Deuterated derivatives of ruxolitinib | |
| US9776973B2 (en) | Deuterated momelotinib | |
| WO2018106916A1 (en) | Deuterated quinoxaline compounds | |
| EP2872159A2 (en) | Deuterated carfilzomib | |
| US10385042B2 (en) | Inhibitors of the enzyme UDP-glucose: N-acyl-sphingosine glucosyltransferase | |
| WO2016109795A1 (en) | Deuterated funapide and difluorofunapide | |
| WO2018013625A1 (en) | Deuterated miconazole | |
| US9676790B2 (en) | Substituted thienotriazolodiazapines | |
| WO2017143038A1 (en) | Deuterated gft-505 | |
| US20140128469A1 (en) | Deuterated n-butyl bumetanide | |
| WO2016061488A1 (en) | Amine reuptake inhibitors | |
| US20180243289A1 (en) | Deuterated morphinan compounds for treating agitation | |
| WO2016105547A1 (en) | Deuterated dasabuvir | |
| WO2014159511A1 (en) | Deuterated pacritinib | |
| KR102433283B1 (en) | Deuterated derivatives of ruxolitinib | |
| EP2542534A1 (en) | Deuterated tetrahydronaphthalene derivatives | |
| WO2010068480A1 (en) | Deuterated derivatives of dimeboline | |
| WO2015009889A1 (en) | Deuterated intedanib derivatives and their use for the treatment of proliferative disorders | |
| KR101900498B1 (en) | Substituted isoindoline-1,3-dione derivatives | |
| WO2018013686A1 (en) | Deuterated idalopirdine | |
| WO2014152275A1 (en) | Deuterium modified derivatives of the ns5b polymerase inhibitor tmc647055 | |
| WO2014150044A1 (en) | Amine reuptake inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15828470 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15828470 Country of ref document: EP Kind code of ref document: A1 |