WO2005028459A1 - Preparation method for quetiapine - Google Patents
Preparation method for quetiapine Download PDFInfo
- Publication number
- WO2005028459A1 WO2005028459A1 PCT/FI2004/000561 FI2004000561W WO2005028459A1 WO 2005028459 A1 WO2005028459 A1 WO 2005028459A1 FI 2004000561 W FI2004000561 W FI 2004000561W WO 2005028459 A1 WO2005028459 A1 WO 2005028459A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- ethyl
- formula
- hydroxyethoxy
- piperazine
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/20—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carbonic acid, or sulfur or nitrogen analogues thereof
- C07D295/215—Radicals derived from nitrogen analogues of carbonic acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D281/00—Heterocyclic compounds containing rings of more than six members having one nitrogen atom and one sulfur atom as the only ring hetero atoms
- C07D281/02—Seven-membered rings
- C07D281/04—Seven-membered rings having the hetero atoms in positions 1 and 4
- C07D281/08—Seven-membered rings having the hetero atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems
- C07D281/12—Seven-membered rings having the hetero atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems condensed with two six-membered rings
- C07D281/16—[b, f]-condensed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention provides an economical alternative method for the preparation of quetiapine in high yield and purity. Further objects of the invention are novel intermediates useful in the process according to the invention.
- quetiapine Several methods for the preparation of quetiapine are known, as disclosed in e.g. GB 8607684, GB 8705574, and WO 01/55125.
- the known methods involve reacting a halo derivative (e.g. iminochloride) of dibenzo[b,f][l,4]-thiazepin-l 1(10- H)-one with l-[2-(hydroxyethoxy)-ethyl]piperazine; reacting the aforementioned halo derivative with piperazine and reacting the resulting intermediate with a haloethoxyethanol; and reacting a haloethylpiperazinylthiazepine derivative with ethylene glycol.
- a halo derivative e.g. iminochloride
- dibenzo[b,f][l,4]-thiazepin-l 1(10- H)-one with l-[2-(hydroxyethoxy)-ethyl]pipe
- the target compound I is obtained by cyclizing a compound of formula II
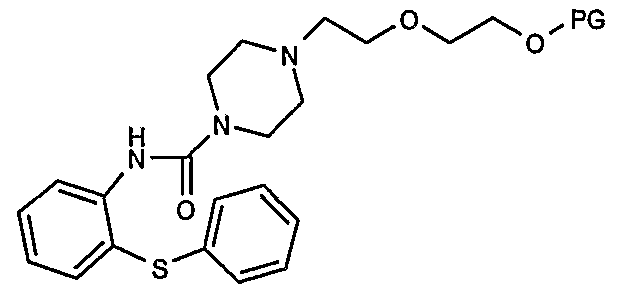
- the compound of formula II is prepared either a) by attaching the protective group PG to the hydroxyl group of compound III
- Ill which may be prepared by a one pot reaction involving 2- phenylsulfanylphenylamine, l-[2-(hydroxyethoxy)-ethyl]piperazine and a coupling agent, e.g. phosgene or equivalent; or
- 2-phenylsulfanylphenylamine maybe prepared e.g. by reacting l-chloro-2- nitrobenzene with benzenethiol and catalytically reducing the nitro group, e.g. as disclosed in the literature. According to the method of the present invention, compound III or IV is obtained without isolation of intermediates by allowing 2- phenylsulfanylphenylamine to react with a carbonyl compound VI
- Rl and R2 may independently be halo, p-nitrophenyl, imidazolyl or -OR wherein R is alkyl or aryl, and adding l-[2-(hydroxyethoxy)-ethyl]piperazine either as such or with a protective group on the hydroxy group.
- Preferred carbonyl compoumds VI include phosgene, diphosgene, triphosgene, (p-nitro)phenylchloroformate, methylchloroformate, dimethyl carbonate and carbonyldi-imidazole.
- Preferred protective groups inlude ethers and esters, e.g. benzoyl, acetyl, benzyl and tetrahydropyryl.
- the reaction of 2-phenylsulfanylphenylamine with the compound of formula VI is preferably carried out in a suitable solvent; preferably toluene, but other aromatic and aliphatic hydrocarbons, also chlorinated derivatives, may be used.
- the reaction temperature may range from - 50 °C to 25 °C.
- the subsequent reaction with protected or unprotected l-[2-(hydroxyethoxy)-ethyl]piperazine is preferably carried out at - 10 °C to 25 °C in the presence of a base, preferably triethylamine but other bases, e.g. other tertiary amines, may be used.
- the protective group PG is subsequently introduced to yield compound II.
- benzoyl chloride is used; other alternatives include acid chlorides and anhydrides, as well as ether-forming reagents.
- the reaction is preferably carried out at a temperature of 0- 100 °C, preferably at ambient temperature.
- Compound TJ is cyclized by treatment with a ring closure agent.
- agents include phosphorus oxychloride, phosphorus pentoxide and polyphosphoric acid.
- An advantageous reagent is a mixture of phosphorus oxychloride and phosphorus pentoxide, preferably using an excess of phosphorus oxychloride as a solvent.
- Possible co-solvents are aliphatic or aromatic hydrocarbons, preferably toluene, as well as chlorinated hydrocarbons.
- the preferable temperature ranges from 50 to 130 °C, preferably about 80 - 100 °C.
- the protective group on the hydroxyl moiety is removed to produce the target compound I, which can be further transferred to a pharmaceutically acceptable salt thereof. If the protective group is susceptible to hydrolysis in basic conditions, sodium hydroxide in ethanol at 20 - 100 °C is preferably used.
- reaction mixture was added to another reaction flask at -10-0 °C, containing the cooled mixture of l-[2-(hydroxyethoxy)-ethyl]- piperazine, triethylamine (2.7 ml) and toluene (20 ml).
- the reaction mixture was stirred at room temperature for 1.5 h.
- Precipitated triethylamine hydrochloride was filtered off.
- the resulting toluene solution was washed twice with saturated NaCl- water (10 ml), dried with K 2 CO 3 and evaporated in vacuo.
Abstract
Description




Claims



Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002538866A CA2538866A1 (en) | 2003-09-23 | 2004-09-23 | Preparation of quetiapine |
| DE602004031129T DE602004031129D1 (en) | 2003-09-23 | 2004-09-23 | PROCESS FOR THE PREPARATION OF QUETIAPIN |
| JP2006526654A JP2007505865A (en) | 2003-09-23 | 2004-09-23 | Manufacture of quetiapine |
| US10/572,370 US7858777B2 (en) | 2003-09-23 | 2004-09-23 | Preparation method for quetiapine |
| AT04767075T ATE496038T1 (en) | 2003-09-23 | 2004-09-23 | METHOD FOR PRODUCING QUETIAPINE |
| EP04767075A EP1664009B1 (en) | 2003-09-23 | 2004-09-23 | Preparation method for quetiapine |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US50498203P | 2003-09-23 | 2003-09-23 | |
| US60/504,982 | 2003-09-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005028459A1 true WO2005028459A1 (en) | 2005-03-31 |
Family
ID=34375549
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FI2004/000561 WO2005028459A1 (en) | 2003-09-23 | 2004-09-23 | Preparation method for quetiapine |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US7858777B2 (en) |
| EP (1) | EP1664009B1 (en) |
| JP (1) | JP2007505865A (en) |
| AT (1) | ATE496038T1 (en) |
| CA (1) | CA2538866A1 (en) |
| DE (1) | DE602004031129D1 (en) |
| WO (1) | WO2005028459A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7687622B2 (en) | 2005-04-14 | 2010-03-30 | Teva Pharmaceutical Industries, Ltd | Process for preparing quetiapine fumarate |
| WO2010100623A1 (en) | 2009-03-04 | 2010-09-10 | Ranbaxy Laboratories Limited | Process for the preparation of quetiapine fumarate |
| US8101597B2 (en) | 2007-05-07 | 2012-01-24 | Actavis Group Ptc Ehf | Quetiapine salts and their polymorphs |
| US8420807B2 (en) | 2008-01-31 | 2013-04-16 | Fermion Oy | Process for the preparation of quetiapine |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003080065A1 (en) * | 2002-03-20 | 2003-10-02 | Teva Pharmaceutical Industries Ltd. | Crystalline forms of quetiapine hemifumarate |
| DE602004024798D1 (en) * | 2003-09-23 | 2010-02-04 | Fermion Oy | PREPARATION OF QUETIAPINE |
| EP2144892A2 (en) * | 2007-03-29 | 2010-01-20 | Teva Pharmaceutical Industries Ltd. | Improved process for preparing quetiapine fumarate |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3539573A (en) * | 1967-03-22 | 1970-11-10 | Jean Schmutz | 11-basic substituted dibenzodiazepines and dibenzothiazepines |
| EP0240228A1 (en) * | 1986-03-27 | 1987-10-07 | Ici Americas Inc. | Thiazepine compounds |
| EP0282236A1 (en) * | 1987-03-10 | 1988-09-14 | Imperial Chemical Industries Plc | Process for the preparation of a thiazepine compound |
| WO2001055125A1 (en) * | 2000-01-25 | 2001-08-02 | EGIS Gyógyszergyár Rt. | A process for the preparation of quetiapine and intermediates therefor |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA979441A (en) * | 1967-02-27 | 1975-12-09 | American Cyanamid Company | 11-(piperazinyl) dibenz (b,f) (1,4) oxazepines and analogous thiazepines |
| US3458516A (en) * | 1968-02-16 | 1969-07-29 | American Cyanamid Co | 11-(piperazinyl)dibenz(b,f)(1,4)oxazepines and analogous thiazepines |
| DE602004024798D1 (en) * | 2003-09-23 | 2010-02-04 | Fermion Oy | PREPARATION OF QUETIAPINE |
-
2004
- 2004-09-23 AT AT04767075T patent/ATE496038T1/en not_active IP Right Cessation
- 2004-09-23 EP EP04767075A patent/EP1664009B1/en active Active
- 2004-09-23 WO PCT/FI2004/000561 patent/WO2005028459A1/en active Application Filing
- 2004-09-23 JP JP2006526654A patent/JP2007505865A/en active Pending
- 2004-09-23 US US10/572,370 patent/US7858777B2/en not_active Expired - Fee Related
- 2004-09-23 CA CA002538866A patent/CA2538866A1/en not_active Abandoned
- 2004-09-23 DE DE602004031129T patent/DE602004031129D1/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3539573A (en) * | 1967-03-22 | 1970-11-10 | Jean Schmutz | 11-basic substituted dibenzodiazepines and dibenzothiazepines |
| EP0240228A1 (en) * | 1986-03-27 | 1987-10-07 | Ici Americas Inc. | Thiazepine compounds |
| EP0282236A1 (en) * | 1987-03-10 | 1988-09-14 | Imperial Chemical Industries Plc | Process for the preparation of a thiazepine compound |
| WO2001055125A1 (en) * | 2000-01-25 | 2001-08-02 | EGIS Gyógyszergyár Rt. | A process for the preparation of quetiapine and intermediates therefor |
Non-Patent Citations (1)
| Title |
|---|
| WARAWA E J ET AL: "Behavioral approach to nondyskinetic dopamine antagonists: Identification of Seroquel", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 44, 1 February 2001 (2001-02-01), pages 372 - 389, XP002213291, ISSN: 0022-2623 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7687622B2 (en) | 2005-04-14 | 2010-03-30 | Teva Pharmaceutical Industries, Ltd | Process for preparing quetiapine fumarate |
| US8101597B2 (en) | 2007-05-07 | 2012-01-24 | Actavis Group Ptc Ehf | Quetiapine salts and their polymorphs |
| US8420807B2 (en) | 2008-01-31 | 2013-04-16 | Fermion Oy | Process for the preparation of quetiapine |
| WO2010100623A1 (en) | 2009-03-04 | 2010-09-10 | Ranbaxy Laboratories Limited | Process for the preparation of quetiapine fumarate |
Also Published As
| Publication number | Publication date |
|---|---|
| ATE496038T1 (en) | 2011-02-15 |
| US7858777B2 (en) | 2010-12-28 |
| DE602004031129D1 (en) | 2011-03-03 |
| JP2007505865A (en) | 2007-03-15 |
| CA2538866A1 (en) | 2005-03-31 |
| US20070111986A1 (en) | 2007-05-17 |
| EP1664009B1 (en) | 2011-01-19 |
| EP1664009A1 (en) | 2006-06-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7863441B2 (en) | Preparation of quetiapine | |
| EP1252151B1 (en) | A process for the preparation of quetiapine and intermediates therefor | |
| EP1963296A2 (en) | Optically active carbamates, process for preparation thereof and use thereof as pharmaceutical intermediates | |
| WO2007020011A1 (en) | Processes for the preparation of thiazepines | |
| EP1664009B1 (en) | Preparation method for quetiapine | |
| WO2007004234A1 (en) | A PROCESS FOR THE PREPARATION OF 2-[2-(4-DIBENZO[b,f] [L,4] THIAZEPIN-11-yl-1- PIPERAZINYL)ETHOXY] ETHANOL FUMARATE | |
| EP2062881B1 (en) | Process for making N-(diphenylmethyl)piperazines | |
| KR100864799B1 (en) | Procedure for preparing 11-4-[2-2-hydroxyethoxyethyl]-1-piperazinyl-dibenzo[b,f][1,4]thiazepine | |
| WO2005028457A1 (en) | Preparation of quetiapine | |
| EP1660469A1 (en) | Process for the preparation of 11-(1-piperazinyl)dibenzo 'b, f! '1 ,4!-thiazepine, an intermediate in the synthesis of the antipsychotic drug quetiapine | |
| AU707950B2 (en) | Heterocyclic compounds for treating myocardial ischemia | |
| WO2009095529A1 (en) | A process for the preparation of quetiapine | |
| US20110172425A1 (en) | Novel water based process for the preparation of substituted diphenylmethyl piperazines | |
| US20110118460A1 (en) | Process for the synthesis of quetiapine | |
| ZA200105600B (en) | Novel process of preparing a benzothiazolone compound. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BW BY BZ CA CH CN CO CR CU CZ DK DM DZ EC EE EG ES FI GB GD GE GM HR HU ID IL IN IS JP KE KG KP KZ LC LK LR LS LT LU LV MA MD MK MN MW MX MZ NA NI NO NZ PG PH PL PT RO RU SC SD SE SG SK SY TJ TM TN TR TT TZ UA UG US UZ VN YU ZA ZM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SZ TZ UG ZM ZW AM AZ BY KG MD RU TJ TM AT BE BG CH CY DE DK EE ES FI FR GB GR HU IE IT MC NL PL PT RO SE SI SK TR BF CF CG CI CM GA GN GQ GW ML MR SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2538866 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004767075 Country of ref document: EP Ref document number: 2006526654 Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004767075 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007111986 Country of ref document: US Ref document number: 10572370 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 10572370 Country of ref document: US |