
Pathophysiology to Risk Factor and Therapeutics to Treatment Strategies on Epilepsy
 , , , , ,
, , , , ,  , and
, and
Abstract
:1. Introduction
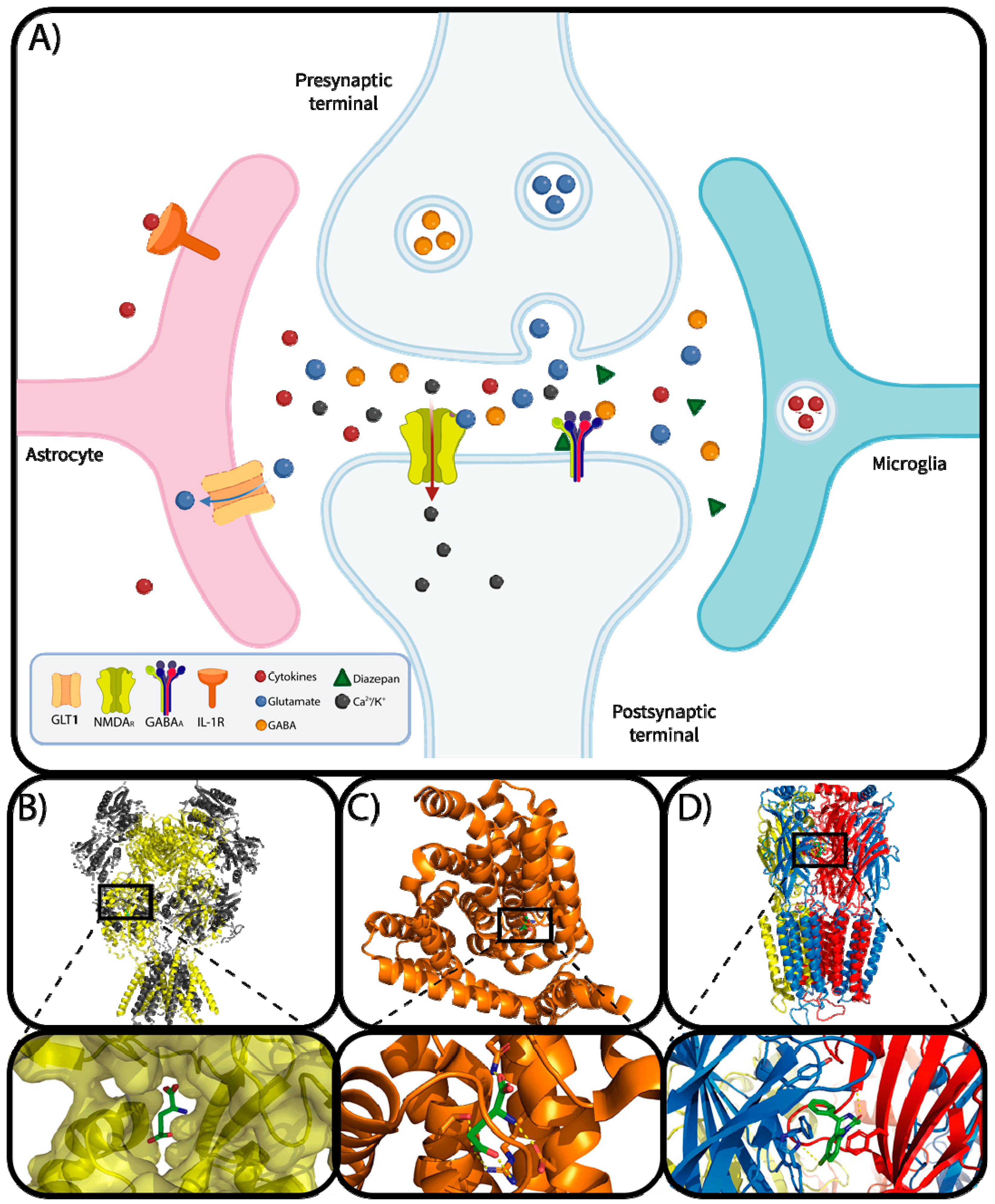
2. Neurological Mechanisms Underlying the Development and Progression of Epilepsy
3. Genetic Influence on Epilepsy
4. New Therapeutic Targets and Pharmacological Strategies
5. Drug Interventions in Epilepsy-Related Clinical Trials
5.1. Lacosamide
5.2. Cannabidiol (CBD)
5.3. Perampanel
5.4. TAK-935
5.5. Ganaxolone
5.6. Everolimus
5.7. Ataluren
5.8. Levetiracetam (LVT) and Valproic Acid (AVP)
5.9. Brivaracetam
5.10. Benzodiazepines
5.11. Lamotrigine (LMT)
5.12. Pregabalin
5.13. Retigabine
5.14. Padsenovil
5.15. Other Clinical Trials
6. Non-Pharmacological Strategies for Epilepsy Treatment
6.1. Ketogenic Diet
Types of Ketogenic Diets
6.2. Neuromodulation Therapy
6.2.1. Invasive
Vagus Nerve Stimulation
Deep Brain Stimulation
Responsive Neural Stimulation
6.2.2. Non-Invasive
Transcranial Magnetic Stimulation and Transcranial Direct Current Stimulation
Ultrasound Stimulation
Transcutaneous VNS
Trigeminal Nerve Stimulation
7. Risk Factors and Comorbidities Related to Epilepsy and AED Treatment
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bertocchi, I.; Cambiaghi, M.; Hasan, M.T. Advances toward precision therapeutics for developmental and epileptic encephalopathies. Front. Neurosci. 2023, 17, 1140679. [Google Scholar] [CrossRef]
- Beghi, E.; Giussani, G.; Sander, J.W. The natural history and prognosis of epilepsy. Epileptic Disord. 2015, 17, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Wirrell, E.; Tinuper, P.; Perucca, E.; Moshé, S.L. Introduction to the epilepsy syndrome papers. Epilepsia 2022, 63, 1330–1332. [Google Scholar] [CrossRef] [PubMed]
- Sirven, J.I. Epilepsy: A Spectrum Disorder. Cold Spring Harb. Perspect. Med. 2015, 5, a022848. [Google Scholar] [CrossRef] [PubMed]
- Guery, D.; Rheims, S. Clinical management of drug resistant epilepsy: A review on current strategies. Neuropsychiatr. Dis. Treat. 2021, 17, 2229–2242. [Google Scholar] [CrossRef]
- Lin, J.J.; Meletti, S.; Vaudano, A.E.; Lin, K.L. Developmental and epileptic encephalopathies: Is prognosis related to different epileptic network dysfunctions? Epilepsy Behav. 2022, 131, 107654. [Google Scholar] [CrossRef]
- Kim, J.E.; Cho, K.O. Functional nutrients for epilepsy. Nutrients 2019, 11, 1309. [Google Scholar] [CrossRef] [PubMed]
- Sarmast, S.T.; Abdullahi, A.M.; Jahan, N. Current Classification of Seizures and Epilepsies: Scope, Limitations and Recommendations for Future Action. Cureus 2020, 12, e10549. [Google Scholar] [CrossRef] [PubMed]
- Manole, A.M.; Sirbu, C.A.; Mititelu, M.R.; Vasiliu, O.; Lorusso, L.; Sirbu, O.M.; Ionita Radu, F. State of the Art and Challenges in Epilepsy—A Narrative Review. J. Pers. Med. 2023, 13, 623. [Google Scholar] [CrossRef]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014, 37, 59–70. [Google Scholar] [CrossRef]
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Estimation of the burden of active and life-time epilepsy: A meta-analytic approach. Epilepsia 2010, 51, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.M.; Henshall, D.C.; Lubin, F.D. The epigenetics of epilepsy and its progression. Neuroscientist 2018, 24, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Aaberg, K.M.; Surén, P.; Søraas, C.L.; Bakken, I.J.; Lossius, M.I.; Stoltenberg, C.; Chin, R. Seizures, syndromes, and etiologies in childhood epilepsy: The International League Against Epilepsy 1981, 1989, and 2017 classifications used in a population-based cohort. Epilepsia 2017, 58, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H.E. The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep. 2007, 7, 348–354. [Google Scholar] [CrossRef]
- Pitkänen, A.; Engel, J. Past and present definitions of epileptogenesis and its biomarkers. Neurotherapeutics 2014, 11, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Alyu, F.; Dikmen, M. Inflammatory aspects of epileptogenesis: Contribution of molecular inflammatory mechanisms. Acta Neuropsychiatr. 2017, 29, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Koh, S. Role of brain inflammation in epileptogenesis. Yonsei Med. J. 2008, 49, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Giaume, C.; Naus, C.C.; Sáez, J.C.; Leybaert, L. Glial Connexins and Pannexins in the Healthy and Diseased Brain. Physiol. Rev. 2021, 101, 93–145. [Google Scholar] [CrossRef] [PubMed]
- Bedner, P.; Steinhäuser, C. Role of Impaired Astrocyte Gap Junction Coupling in Epileptogenesis. Cells 2023, 12, 1669. [Google Scholar] [CrossRef] [PubMed]
- Hardy, E.; Moulard, J.; Walter, A.; Ezan, P.; Bemelmans, A.-P.; Mouthon, F.; Charvériat, M.; Rouach, N.; Rancillac, A. Upregulation of astroglial connexin 30 impairs hippocampal synaptic activity and recognition memory. PLoS Biol. 2023, 21, e3002075. [Google Scholar] [CrossRef]
- Victor, T.R.; Tsirka, S.E. Microglial contributions to aberrant neurogenesis and pathophysiology of epilepsy. Neuroimmunol. Neuroinflamm. 2020, 7, 234–247. [Google Scholar] [CrossRef]
- Kinoshita, S.; Koyama, R. Pro- and anti-epileptic roles of microglia. Neural Regen. Res. 2021, 16, 1369–1371. [Google Scholar] [CrossRef]
- Scorza, C.A.; Marques, M.J.G.; da Silva, S.G.; da Graça Naffah-Mazzacoratti, M.; Scorza, F.A.; Cavalheiro, E.A. Status epilepticus does not induce acute brain inflammatory response in the Amazon rodent Proechimys, an animal model resistant to epileptogenesis. Neurosci. Lett. 2018, 668, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Hanke, M.L.; Kielian, T. Toll-like receptors in health and disease in the brain: Mechanisms and therapeutic potential. Clin. Sci. 2011, 121, 367–387. [Google Scholar] [CrossRef] [PubMed]
- Cimino, P.J.; Keene, C.D.; Breyer, R.M.; Montine, K.S.; Montine, T.J. Therapeutic targets in prostaglandin E2 signaling for neurologic disease. Curr. Med. Chem. 2008, 15, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-S.; Huang, T.-H.; Lai, M.-C.; Huang, C.-W. The Role of Glutamate Receptors in Epilepsy. Biomedicines 2023, 11, 783. [Google Scholar] [CrossRef]
- Barker-Haliski, M.; White, H.S. Glutamatergic Mechanisms Associated with Seizures and Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022863. [Google Scholar] [CrossRef]
- Sarlo, G.L.; Holton, K.F. Brain concentrations of glutamate and GABA in human epilepsy: A review. Seizure 2021, 91, 213–227. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef]
- Rowley, N.M.; Madsen, K.K.; Schousboe, A.; Steve White, H. Glutamate and GABA synthesis, release, transport and metabolism as targets for seizure control. Neurochem. Int. 2012, 61, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Simonato, M. Epilepsy an update on disease mechanisms: The potential role of microRNAs. Front. Neurol. 2018, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T. Ionotropic Glutamate Receptors in Epilepsy: A Review Focusing on AMPA and NMDA Receptors. Biomolecules 2020, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, M.; Mefford, H.C. Recent advances in epilepsy genomics and genetic testing. F1000Research 2020, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.L.; Lauerer, R.J.; Bahr, J.C.; Souza, I.A.; Myers, C.T.; Uysal, B.; Schwarz, N.; Gandini, M.A.; Huang, S.; Keren, B.; et al. De Novo Pathogenic Variants in CACNA1E Cause Developmental and Epileptic Encephalopathy with Contractures, Macrocephaly, and Dyskinesias. Am. J. Hum. Genet. 2018, 103, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Symonds, J.D.; Zuberi, S.M.; Johnson, M.R. Advances in epilepsy gene discovery and implications for epilepsy diagnosis and treatment. Curr. Opin. Neurol. 2017, 30, 193–199. [Google Scholar] [CrossRef]
- Masnada, S.; Hedrich, U.B.S.; Gardella, E.; Schubert, J.; Kaiwar, C.; Klee, E.W.; Lanpher, B.C.; Gavrilova, R.H.; Synofzik, M.; Bast, T.; et al. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain 2017, 140, 2337–2354. [Google Scholar] [CrossRef]
- Samanta, D. DEPDC5-related epilepsy: A comprehensive review. Epilepsy Behav. 2022, 130, 108678. [Google Scholar] [CrossRef]
- Rochtus, A.; Olson, H.E.; Smith, L.; Keith, L.G.; El Achkar, C.; Taylor, A.; Mahida, S.; Park, M.; Kelly, M.; Shain, C.; et al. Genetic diagnoses in epilepsy: The impact of dynamic exome analysis in a pediatric cohort. Epilepsia 2020, 61, 249–258. [Google Scholar] [CrossRef]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef]
- Kalilani, L.; Sun, X.; Pelgrims, B.; Noack-Rink, M.; Villanueva, V. The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia 2018, 59, 2179–2193. [Google Scholar] [CrossRef] [PubMed]
- Raut, D.; Bhatt, L.K. Evolving targets for anti-epileptic drug discovery. Eur. J. Pharmacol. 2020, 887, 173582. [Google Scholar] [CrossRef] [PubMed]
- Stefanescu, R.A.; Shivakeshavan, R.G.; Talathi, S.S. Computational models of epilepsy. Seizure 2012, 21, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Xu, H.; Ma, H.; Luo, L.; Yang, L.; Chen, F.; Qu, X.; Liu, H.; Zhang, R. LncRNA CASC2 inhibits astrocytic activation and adenosine metabolism by regulating PTEN in pentylenetetrazol-induced epilepsy model. J. Chem. Neuroanat. 2020, 105, 101749. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, A.; DuPont, S.; Zhou, D.; Matta, M.; Nagire, V.; Lagae, L. Current role of carbamazepine and oxcarbazepine in the management of epilepsy. Seizure 2020, 83, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, Y.C.; Demir, O. Use of Phenytoin, Phenobarbital Carbamazepine, Levetiracetam Lamotrigine and Valproate in Pregnancy and Breastfeeding: Risk of Major Malformations, Dose-dependency, Monotherapy vs Polytherapy, Pharmacokinetics and Clinical Implications. Curr. Neuropharmacol. 2021, 19, 1805–1824. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-Y.; Wang, H.-Y.; Wen, B.; Yang, Z.-B.; Feng, K.; Fan, J.-C. A Comparison of Midazolam, Lorazepam, and Diazepam for the Treatment of Status Epilepticus in Children: A Network Meta-analysis. J. Child Neurol. 2016, 31, 1093–1107. [Google Scholar] [CrossRef]
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef]
- Nevitt, S.J.; Sudell, M.; Cividini, S.; Marson, A.G.; Tudur Smith, C. Antiepileptic drug monotherapy for epilepsy: A network meta-analysis of individual participant data. Cochrane Database Syst. Rev. 2022, 4, CD011412. [Google Scholar] [CrossRef]
- Niquet, J.; Nguyen, D.; de Araujo Furtado, M.; Lumley, L. Treatment of cholinergic-induced status epilepticus with polytherapy targeting GABA and glutamate receptors. Epilepsia Open 2023, 8 (Suppl. S1), S117–S140. [Google Scholar] [CrossRef]
- Verrotti, A.; Lattanzi, S.; Brigo, F.; Zaccara, G. Pharmacodynamic interactions of antiepileptic drugs: From bench to clinical practice. Epilepsy Behav. 2020, 104, 106939. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.J.; Pitkänen, A. Biomarkers for epileptogenesis and its treatment. Neuropharmacology 2020, 167, 107735. [Google Scholar] [CrossRef] [PubMed]
- Noam, Y.; Raol, Y.H.; Holmes, G.L. Searching for new targets for treatment of pediatric epilepsy. Epilepsy Behav. 2013, 26, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Conboy, K.; Henshall, D.C.; Brennan, G.P. Epigenetic principles underlying epileptogenesis and epilepsy syndromes. Neurobiol. Dis. 2021, 148, 105179. [Google Scholar] [CrossRef] [PubMed]
- Soltani Khaboushan, A.; Yazdanpanah, N.; Rezaei, N. Neuroinflammation and Proinflammatory Cytokines in Epileptogenesis. Mol. Neurobiol. 2022, 59, 1724–1743. [Google Scholar] [CrossRef]
- Lagarde, S.; Villeneuve, N.; Trébuchon, A.; Kaphan, E.; Lepine, A.; McGonigal, A.; Roubertie, A.; Barthez, M.-A.J.; Trommsdorff, V.; Lefranc, J.; et al. Anti-tumor necrosis factor alpha therapy (adalimumab) in Rasmussen’s encephalitis: An open pilot study. Epilepsia 2016, 57, 956–966. [Google Scholar] [CrossRef]
- Leo, A.; Nesci, V.; Tallarico, M.; Amodio, N.; Gallo Cantafio, E.M.; De Sarro, G.; Constanti, A.; Russo, E.; Citraro, R. IL-6 Receptor Blockade by Tocilizumab Has Anti-absence and Anti-epileptogenic Effects in the WAG/Rij Rat Model of Absence Epilepsy. Neurother. J. Am. Soc. Exp. Neurother. 2020, 17, 2004–2014. [Google Scholar] [CrossRef]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef]
- Ravizza, T.; Lucas, S.-M.; Balosso, S.; Bernardino, L.; Ku, G.; Noé, F.; Malva, J.; Randle, J.C.R.; Allan, S.; Vezzani, A. Inactivation of caspase-1 in rodent brain: A novel anticonvulsive strategy. Epilepsia 2006, 47, 1160–1168. [Google Scholar] [CrossRef]
- Williams, S.; Hamil, N.; Abramov, A.Y.; Walker, M.C.; Kovac, S. Status epilepticus results in persistent overproduction of reactive oxygen species, inhibition of which is neuroprotective. Neuroscience 2015, 303, 160–165. [Google Scholar] [CrossRef]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.M.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Musto, A.E. The role of inflammation in the development of epilepsy. J. Neuroinflamm. 2018, 15, 144. [Google Scholar] [CrossRef] [PubMed]
- Terrone, G.; Salamone, A.; Vezzani, A. Inflammation and Epilepsy: Preclinical Findings and Potential Clinical Translation. Curr. Pharm. Des. 2017, 23, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Bauer, S.; Bozzi, Y.; Caleo, M.; Dingledine, R.; Gorter, J.A.; Henshall, D.C.; Kaufer, D.; Koh, S.; Löscher, W.; et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia 2017, 58 (Suppl. S3), 27–38. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Deng, X.-J.; Xu, D. Microglia in epilepsy. Neurobiol. Dis. 2023, 185, 106249. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef]
- Akyuz, E.; Polat, A.K.; Eroglu, E.; Kullu, I.; Angelopoulou, E.; Paudel, Y.N. Revisiting the role of neurotransmitters in epilepsy: An updated review. Life Sci. 2021, 265, 118826. [Google Scholar] [CrossRef]
- Dejakaisaya, H.; Kwan, P.; Jones, N.C. Astrocyte and glutamate involvement in the pathogenesis of epilepsy in Alzheimer’s disease. Epilepsia 2021, 62, 1485–1493. [Google Scholar] [CrossRef]
- Sühs, K.-W.; Gudi, V.; Eckermann, N.; Fairless, R.; Pul, R.; Skripuletz, T.; Stangel, M. Cytokine regulation by modulation of the NMDA receptor on astrocytes. Neurosci. Lett. 2016, 629, 227–233. [Google Scholar] [CrossRef]
- Aloisi, F.; Borsellino, G.; Caré, A.; Testa, U.; Gallo, P.; Russo, G.; Peschle, C.; Levi, G. Cytokine regulation of astrocyte function: In-vitro studies using cells from the human brain. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 1995, 13, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Prow, N.A.; Irani, D.N. The inflammatory cytokine, interleukin-1 beta, mediates loss of astroglial glutamate transport and drives excitotoxic motor neuron injury in the spinal cord during acute viral encephalomyelitis. J. Neurochem. 2008, 105, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Zumkehr, J.; Rodriguez-Ortiz, C.J.; Medeiros, R.; Kitazawa, M. Inflammatory Cytokine, IL-1β, Regulates Glial Glutamate Transporter via microRNA-181a in vitro. J. Alzheimer’s Dis. 2018, 63, 965–975. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.A.; Aronica, E.; Vezzani, A.; Ravizza, T. Review: Neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: Emerging evidence from preclinical and clinical studies. Neuropathol. Appl. Neurobiol. 2018, 44, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, H.; Geng, Z.; Yu, Z.; Li, H.; Dong, Y.; Zhang, H.; Huang, Z.; Jiang, J.; Zhao, Y. Structural basis of ligand binding modes of human EAAT2. Nat. Commun. 2022, 13, 3329. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Shan, W.; Yang, H.; Liu, R.; Wu, J.; Wang, Q. The Role of Neuroinflammation in Post-traumatic Epilepsy. Front. Neurol. 2021, 12, 646152. [Google Scholar] [CrossRef]
- Cerne, R.; Lippa, A.; Poe, M.M.; Smith, J.L.; Jin, X.; Ping, X.; Golani, L.K.; Cook, J.M.; Witkin, J.M. GABAkines—Advances in the discovery, development, and commercialization of positive allosteric modulators of GABA(A) receptors. Pharmacol. Ther. 2022, 234, 108035. [Google Scholar] [CrossRef]
- Tong, X.; Zhang, Z.; Zhu, J.; Li, S.; Qu, S.; Qin, B.; Guo, Y.; Chen, R. A Comparison of Epileptogenic Effect of Status Epilepticus Treated with Diazepam, Midazolam, and Pentobarbital in the Mouse Pilocarpine Model of Epilepsy. Front. Neurol. 2022, 13, 821917. [Google Scholar] [CrossRef]
- Masiulis, S.; Desai, R.; Uchański, T.; Serna Martin, I.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABA(A) receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- de Araújo Boleti, A.P.; de Oliveira Flores, T.M.; Moreno, S.E.; Anjos, L.D.; Mortari, M.R.; Migliolo, L. Neuroinflammation: An overview of neurodegenerative and metabolic diseases and of biotechnological studies. Neurochem. Int. 2020, 136, 104714. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, P.H.d.O.; Boleti, A.P.d.A.; Silva, P.S.e.; Mukoyama, L.T.H.; Guindo, A.S.; Moraes, L.F.R.N.d.; Oliveira, C.F.R.d.; Macedo, M.L.R.; Carvalho, C.M.E.; de Castro, A.P.; et al. Evaluation of a Novel Synthetic Peptide Derived from Cytolytic Mycotoxin Candidalysin. Toxins 2022, 14, 696. [Google Scholar] [CrossRef] [PubMed]
- Janković, S.M.; Đešević, M. Advancements in neuroactive peptides in seizures. Expert Rev. Neurother. 2022, 22, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Li, X.; Du, H.; Jiang, C.; Xu, S.; Cao, Y. Immunomodulatory significance of natural peptides in mammalians: Promising agents for medical application. Immunobiology 2020, 225, 151936. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Absalom, N.; Biggs, K.; King, G.F.; Ma, L. Venom-derived modulators of epilepsy-related ion channels. Biochem. Pharmacol. 2020, 181, 114043. [Google Scholar] [CrossRef] [PubMed]
- de Moraes, L.F.R.N.; Silva, P.S.E.; Pereira, T.C.P.L.; Almeida Rodrigues, T.A.; Farias Frihling, B.E.; da Costa, R.A.; Torquato, H.F.V.; Lima, C.S.; Paredes-Gamero, E.J.; Migliolo, L. First generation of multifunctional peptides derived from latarcin-3a from Lachesana tarabaevi spider toxin. Front. Microbiol. 2022, 13, 965621. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Zhu, L.; Wang, L.; Liu, X.; Xiao, F.; Xie, Y.; Zheng, W.; Jiang, X. Screening on-chip fabricated nanoparticles for penetrating the blood-brain barrier. Nanoscale 2022, 14, 3234–3241. [Google Scholar] [CrossRef]
- Edwards, A.B.; Mastaglia, F.L.; Knuckey, N.W.; Meloni, B.P. Neuroprotective Cationic Arginine-Rich Peptides (CARPs): An Assessment of Their Clinical Safety. Drug Saf. 2020, 43, 957–969. [Google Scholar] [CrossRef]
- Boleti, A.P.A.; Frihling, B.E.F.; ESilva, P.S.; Cardoso, P.H.O.; de Moraes, L.F.R.N.; Rodrigues, T.A.A.; Biembengute, M.E.F.; Koolen, H.H.F.; Migliolo, L. Biochemical aspects and therapeutic mechanisms of cannabidiol in epilepsy. Neurosci. Biobehav. Rev. 2022, 132, 1214–1228. [Google Scholar] [CrossRef]
- Janz, P.; Hauser, P.; Heining, K.; Nestel, S.; Kirsch, M.; Egert, U.; Haas, C.A. Position- and Time-Dependent Arc Expression Links Neuronal Activity to Synaptic Plasticity during Epileptogenesis. Front. Cell. Neurosci. 2018, 12, 244. [Google Scholar] [CrossRef]
- de Oliveira, R.M.W. Neuroplasticity. J. Chem. Neuroanat. 2020, 108, 101822. [Google Scholar] [CrossRef] [PubMed]
- Sibarov, D.A.; Tsytsarev, V.; Volnova, A.; Vaganova, A.N.; Alves, J.; Rojas, L.; Sanabria, P.; Ignashchenkova, A.; Savage, E.D.; Inyushin, M. Arc protein, a remnant of ancient retrovirus, forms virus-like particles, which are abundantly generated by neurons during epileptic seizures, and affects epileptic susceptibility in rodent models. Front. Neurol. 2023, 14, 1201104. [Google Scholar] [CrossRef] [PubMed]
- Rosi, S. Neuroinflammation and the plasticity-related immediate-early gene Arc. Brain Behav. Immun. 2011, 25 (Suppl. S1), S39–S49. [Google Scholar] [CrossRef] [PubMed]
- Pong, A.W.; Ross, J.; Tyrlikova, I.; Giermek, A.J.; Kohli, M.P.; Khan, Y.A.; Salgado, R.D.; Klein, P. Epilepsy: Expert opinion on emerging drugs in phase 2/3 clinical trials. Expert Opin. Emerg. Drugs 2022, 27, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Umscheid, C.A.; Margolis, D.J.; Grossman, C.E. Key concepts of clinical trials: A narrative review. Postgrad. Med. 2011, 123, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Minneci, P.C.; Deans, K.J. Clinical trials. Semin. Pediatr. Surg. 2018, 27, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Carona, A.; Bicker, J.; Silva, R.; Fonseca, C.; Falcão, A.; Fortuna, A. Pharmacology of lacosamide: From its molecular mechanisms and pharmacokinetics to future therapeutic applications. Life Sci. 2021, 275, 119342. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Tofighy, A.; White, H.S.; Matagne, A.; Wolff, C. Current understanding of the mechanism of action of the antiepileptic drug lacosamide. Epilepsy Res. 2015, 110, 189–205. [Google Scholar] [CrossRef]
- Beydoun, A.; D’Souza, J.; Hebert, D.; Doty, P. Lacosamide: Pharmacology, mechanisms of action and pooled efficacy and safety data in partial-onset seizures. Expert Rev. Neurother. 2009, 9, 33–42. [Google Scholar] [CrossRef]
- Gray, R.A.; Whalley, B.J. The proposed mechanisms of action of CBD in epilepsy. Epileptic Disord. 2020, 22, 10–15. [Google Scholar] [CrossRef]
- Szaflarski, J.P.; Hernando, K.; Bebin, E.M.; Gaston, T.E.; Grayson, L.E.; Ampah, S.B.; Moreadith, R. Higher cannabidiol plasma levels are associated with better seizure response following treatment with a pharmaceutical grade cannabidiol. Epilepsy Behav. 2019, 95, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Halford, J.J.; Miller, I.; Nabbout, R.; Sanchez-Carpintero, R.; Shiloh-Malawsky, Y.; Wong, M.; Zolnowska, M.; Checketts, D.; Dunayevich, E.; et al. Add-on cannabidiol in patients with Dravet syndrome: Results of a long-term open-label extension trial. Epilepsia 2021, 62, 2505–2517. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.D.; Mazurkiewicz-Bełdzińska, M.; Chin, R.F.; Gil-Nagel, A.; Gunning, B.; Halford, J.J.; Mitchell, W.; Scott Perry, M.; Thiele, E.A.; Weinstock, A.; et al. Long-term safety and efficacy of add-on cannabidiol in patients with Lennox-Gastaut syndrome: Results of a long-term open-label extension trial. Epilepsia 2021, 62, 2228–2239. [Google Scholar] [CrossRef] [PubMed]
- Franco, V.; Crema, F.; Iudice, A.; Zaccara, G.; Grillo, E. Novel treatment options for epilepsy: Focus on perampanel. Pharmacol. Res. 2013, 70, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Hashizume, Y.; Tokuhara, N.; Takenaka, O.; Kohmura, N.; Ogasawara, A.; Hatakeyama, S.; Ohgoh, M.; Ueno, M.; Nishizawa, Y. Perampanel: A novel, orally active, noncompetitive AMPA-receptor antagonist that reduces seizure activity in rodent models of epilepsy. Epilepsia 2011, 52, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Gil-Nagel, A.; Wheless, J.W.; Kim, J.H.; Wechsler, R.T. Perampanel monotherapy for the treatment of epilepsy: Clinical trial and real-world evidence. Epilepsy Behav. 2022, 136, 108885. [Google Scholar] [CrossRef] [PubMed]
- Fogarasi, A.; Flamini, R.; Milh, M.; Phillips, S.; Yoshitomi, S.; Patten, A.; Takase, T.; Laurenza, A.; Ngo, L.Y. Open-label study to investigate the safety and efficacy of adjunctive perampanel in pediatric patients (4 to <12 years) with inadequately controlled focal seizures or generalized tonic-clonic seizures. Epilepsia 2020, 61, 125–137. [Google Scholar] [CrossRef]
- Trigg, A.; Brohan, E.; Cocks, K.; Jones, A.; Tahami Monfared, A.A.; Chabot, I.; Meier, G.; Campbell, R.; Li, H.; Ngo, L.Y. Health-related quality of life in pediatric patients with partial onset seizures or primary generalized tonic-clonic seizures receiving adjunctive perampanel. Epilepsy Behav. 2021, 118, 107938. [Google Scholar] [CrossRef]
- Koike, T.; Yoshikawa, M.; Ando, H.K.; Farnaby, W.; Nishi, T.; Watanabe, E.; Yano, J.; Miyamoto, M.; Kondo, S.; Ishii, T.; et al. Discovery of Soticlestat, a Potent and Selective Inhibitor for Cholesterol 24-Hydroxylase (CH24H). J. Med. Chem. 2021, 64, 12228–12244. [Google Scholar] [CrossRef]
- Nishi, T.; Kondo, S.; Miyamoto, M.; Watanabe, S.; Hasegawa, S.; Kondo, S.; Yano, J.; Watanabe, E.; Ishi, T.; Yoshikawa, M.; et al. Soticlestat, a novel cholesterol 24-hydroxylase inhibitor shows a therapeutic potential for neural hyperexcitation in mice. Sci. Rep. 2020, 10, 17081. [Google Scholar] [CrossRef]
- Hahn, C.D.; Jiang, Y.; Villanueva, V.; Zolnowska, M.; Arkilo, D.; Hsiao, S.; Asgharnejad, M.; Dlugos, D. A phase 2, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of soticlestat as adjunctive therapy in pediatric patients with Dravet syndrome or Lennox-Gastaut syndrome (ELEKTRA). Epilepsia 2022, 63, 2671–2683. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Ganaxolone: First Approval. Drugs 2022, 82, 933–940. [Google Scholar] [CrossRef]
- Lattanzi, S.; Riva, A.; Striano, P. Ganaxolone treatment for epilepsy patients: From pharmacology to place in therapy. Expert Rev. Neurother. 2021, 21, 1317–1332. [Google Scholar] [CrossRef] [PubMed]
- Yasmen, N.; Sluter, M.N.; Yu, Y.; Jiang, J. Ganaxolone for management of seizures associated with CDKL5 deficiency disorder. Trends Pharmacol. Sci. 2023, 44, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.B.; Wood, P.L.; Wieland, S.; Hawkinson, J.E.; Belelli, D.; Lambert, J.J.; White, H.S.; Wolf, H.H.; Mirsadeghi, S.; Tahir, S.H.; et al. Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3alpha-hydroxy-3beta-methyl-5alpha-pregnan-20-one), a selective, high-affinity, steroid modulator of the gamma-aminobutyric acid(A) receptor. J. Pharmacol. Exp. Ther. 1997, 280, 1284–1295. [Google Scholar] [PubMed]
- French, J.A.; Lawson, J.A.; Yapici, Z.; Ikeda, H.; Polster, T.; Nabbout, R.; Curatolo, P.; de Vries, P.J.; Dlugos, D.J.; Berkowitz, N. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): A phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016, 388, 2153–2163. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; King, L.; Bluvstein, J.; Friedman, D. Ataluren for drug-resistant epilepsy in nonsense variant-mediated Dravet syndrome and CDKL5 deficiency disorder. Ann. Clin. Transl. Neurol. 2021, 8, 639–644. [Google Scholar] [CrossRef]
- Abou-Khalil, B. Levetiracetam in the treatment of epilepsy. Neuropsychiatr. Dis. Treat. 2008, 4, 507–523. [Google Scholar] [CrossRef]
- Madeo, M.; Kovács, A.D.; Pearce, D.A. The Human Synaptic Vesicle Protein, SV2A, Functions as a Galactose Transporter in Saccharomyces cerevisiae. J. Biol. Chem. 2014, 289, 33066–33071. [Google Scholar] [CrossRef]
- Lloyd, K.A. A scientific review: Mechanisms of valproate-mediated teratogenesis. Biosci. Horizons 2013, 6, hzt003. [Google Scholar] [CrossRef]
- Maan, J.S.; Duong, T.V.H.; Saadabadi, A. Carbamazepine. 2018. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482455/ (accessed on 15 August 2023).
- Griffin, C.E.; Kaye, A.M.; Bueno, F.R.; Kaye, A.D. Benzodiazepine pharmacology and central nervous system–mediated effects. Ochsner J. 2013, 13, 214–223. [Google Scholar] [PubMed]
- Seo, H.-J.; Chiesa, A.; Lee, S.-J.; Patkar, A.A.; Han, C.; Masand, P.S.; Serretti, A.; Pae, C.-U. Safety and tolerability of lamotrigine: Results from 12 placebo-controlled clinical trials and clinical implications. Clin. Neuropharmacol. 2011, 34, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Micó, J.-A.; Prieto, R. Elucidating the mechanism of action of pregabalin: α2δ as a therapeutic target in anxiety. CNS Drugs 2012, 26, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Gunthorpe, M.J.; Large, C.H.; Sankar, R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 2012, 53, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A. Antiseizure medication discovery: Recent and future paradigm shifts. Epilepsia Open 2022, 7, S133–S141. [Google Scholar] [CrossRef]
- Boddu, S.H.S.; Kumari, S. A short review on the intranasal delivery of diazepam for treating acute repetitive seizures. Pharmaceutics 2020, 12, 1167. [Google Scholar] [CrossRef]
- Spencer, D.C.; Sinha, S.R.; Choi, E.J.; Cleveland, J.M.; King, A.; Meng, T.; Pullman, W.E.; Sequeira, D.J.; Van Ess, P.J.; Wheless, J.W. Safety and efficacy of midazolam nasal spray for the treatment of intermittent bouts of increased seizure activity in the epilepsy monitoring unit: A double-blind, randomized, placebo-controlled trial. Epilepsia 2020, 61, 2415–2425. [Google Scholar] [CrossRef]
- Nabbout, R.; Mistry, A.; Zuberi, S.; Villeneuve, N.; Gil-Nagel, A.; Sanchez-Carpintero, R.; Stephani, U.; Laux, L.; Wirrell, E.; Knupp, K. Fenfluramine for treatment-resistant seizures in patients with Dravet syndrome receiving stiripentol-inclusive regimens: A randomized clinical trial. JAMA Neurol. 2020, 77, 300–308. [Google Scholar] [CrossRef]
- Schoonjans, A.-S.; Ceulemans, B. A critical evaluation of fenfluramine hydrochloride for the treatment of Dravet syndrome. Expert Rev. Neurother. 2022, 22, 351–364. [Google Scholar] [CrossRef]
- Sullivan, J.; Specchio, N.; Devinsky, O.; Auvin, S.; Perry, M.S.; Strzelczyk, A.; Gil-Nagel, A.; Dai, D.; Galer, B.S.; Gammaitoni, A.R. Fenfluramine significantly reduces day-to-day seizure burden by increasing number of seizure-free days and time between seizures in patients with Dravet syndrome: A time-to-event analysis. Epilepsia 2022, 63, 130–138. [Google Scholar] [CrossRef]
- Zhu, H.; Bi, D.; Zhang, Y.; Kong, C.; Du, J.; Wu, X.; Wei, Q.; Qin, H. Ketogenic diet for human diseases: The underlying mechanisms and potential for clinical implementations. Signal Transduct. Target. Ther. 2022, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, L.P.d.B. Ketogenic diet for epilepsy treatment. Arq. Neuro-Psiquiatria 2016, 74, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Sondhi, V.; Agarwala, A.; Pandey, R.M.; Chakrabarty, B.; Jauhari, P.; Lodha, R.; Toteja, G.S.; Sharma, S.; Paul, V.K.; Kossoff, E.; et al. Efficacy of Ketogenic Diet, Modified Atkins Diet, and Low Glycemic Index Therapy Diet Among Children With Drug-Resistant Epilepsy: A Randomized Clinical Trial. JAMA Pediatr. 2020, 174, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Barzegar, M.; Afghan, M.; Tarmahi, V.; Behtari, M.; Rahimi Khamaneh, S.; Raeisi, S. Ketogenic diet: Overview, types, and possible anti-seizure mechanisms. Nutr. Neurosci. 2021, 24, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Han, F.Y.; Conboy-Schmidt, L.; Rybachuk, G.; Volk, H.A.; Zanghi, B.; Pan, Y.; Borges, K. Dietary medium chain triglycerides for management of epilepsy: New data from human, dog, and rodent studies. Epilepsia 2021, 62, 1790–1806. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, L.; Li, H.; Liu, Y. Effect of low glycaemic diet and structured exercise on quality of life and psychosocial functions in children with epilepsy. J. Int. Med. Res. 2020, 48, 300060519893855. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Ketogenic Diet and Epilepsy. Nutrients 2019, 11, 2510. [Google Scholar] [CrossRef]
- Abouelleil, M.; Deshpande, N.; Ali, R. Emerging trends in neuromodulation for treatment of drug-resistant epilepsy. Front. Pain Res. 2022, 3, 839463. [Google Scholar] [CrossRef]
- Parihar, J.; Agrawal, M.; Samala, R.; Chandra, P.S.; Tripathi, M. Role of neuromodulation for treatment of drug-resistant epilepsy. Neurol. India 2020, 68, 249. [Google Scholar]
- Nune, G.; DeGiorgio, C.; Heck, C. Neuromodulation in the treatment of epilepsy. Curr. Treat. Opt. Neurol. 2015, 17, 1–6. [Google Scholar] [CrossRef]
- Davis, P.; Gaitanis, J. Neuromodulation for the treatment of epilepsy: A review of current approaches and future directions. Clin. Ther. 2020, 42, 1140–1154. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, L.; Xu, C.; Chen, Z.; Wang, Y. Non-invasive sensory neuromodulation in epilepsy: Updates and future perspectives. Neurobiol. Dis. 2023, 179, 106049. [Google Scholar] [CrossRef] [PubMed]
- Howland, R.H. Vagus nerve stimulation. Curr. Behav. Neurosci. Rep. 2014, 1, 64–73. [Google Scholar] [CrossRef]
- Johnson, R.L.; Wilson, C.G. A review of vagus nerve stimulation as a therapeutic intervention. J. Inflamm. Res. 2018, 11, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Ekmekçi, H.; Kaptan, H. Vagus nerve stimulation. Open Access Maced. J. Med. Sci. 2017, 5, 391. [Google Scholar] [CrossRef] [PubMed]
- Wheless, J.W.; Gienapp, A.J.; Ryvlin, P. Vagus nerve stimulation (VNS) therapy update. Epilepsy Behav. 2018, 88, 2–10. [Google Scholar] [CrossRef]
- Zangiabadi, N.; Ladino, L.D.; Sina, F.; Orozco-Hernández, J.P.; Carter, A.; Téllez-Zenteno, J.F. Deep Brain Stimulation and Drug-Resistant Epilepsy: A Review of the Literature. Front. Neurol. 2019, 10, 601. [Google Scholar] [CrossRef]
- Lega, B.C.; Halpern, C.H.; Jaggi, J.L.; Baltuch, G.H. Deep brain stimulation in the treatment of refractory epilepsy: Update on current data and future directions. Neurobiol. Dis. 2010, 38, 354–360. [Google Scholar] [CrossRef]
- Klinger, N.; Mittal, S. Deep brain stimulation for seizure control in drug-resistant epilepsy. Neurosurg. Focus 2018, 45, E4. [Google Scholar] [CrossRef]
- Salanova, V. Deep brain stimulation for epilepsy. Epilepsy Behav. 2018, 88, 21–24. [Google Scholar] [CrossRef]
- Morrell, M.J.; Halpern, C. Responsive direct brain stimulation for epilepsy. Neurosurg. Clin. 2016, 27, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Carrette, S.; Boon, P.; Sprengers, M.; Raedt, R.; Vonck, K. Responsive neurostimulation in epilepsy. Expert Rev. Neurother. 2015, 15, 1445–1454. [Google Scholar] [CrossRef]
- Chen, R.; Spencer, D.C.; Weston, J.; Nolan, S.J. Transcranial magnetic stimulation for the treatment of epilepsy. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tsuboyama, M.; Kaye, H.L.; Rotenberg, A. Review of transcranial magnetic stimulation in epilepsy. Clin. Ther. 2020, 42, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.S.; Müller, V.T.; da Mota Gomes, M.; Rotenberg, A.; Fregni, F. Safety of repetitive transcranial magnetic stimulation in patients with epilepsy: A systematic review. Epilepsy Behav. 2016, 57, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.-S.; Ripa, V.; Al-Awar, O.; Panov, F.; Ghatan, S.; Jetté, N. Epilepsy and neuromodulation—Randomized controlled trials. Brain Sci. 2018, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Hakimova, H.; Kim, S.; Chu, K.; Lee, S.K.; Jeong, B.; Jeon, D. Ultrasound stimulation inhibits recurrent seizures and improves behavioral outcome in an experimental model of mesial temporal lobe epilepsy. Epilepsy Behav. 2015, 49, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.; Yan, J.; Wang, X.; Yuan, Y.; Li, X. Seizure control by low-intensity ultrasound in mice with temporal lobe epilepsy. Epilepsy Res. 2019, 154, 1–7. [Google Scholar] [CrossRef]
- Bauer, S.; Baier, H.; Baumgartner, C.; Bohlmann, K.; Fauser, S.; Graf, W.; Hillenbrand, B.; Hirsch, M.; Last, C.; Lerche, H. Transcutaneous vagus nerve stimulation (tVNS) for treatment of drug-resistant epilepsy: A randomized, double-blind clinical trial (cMPsE02). Brain Stimul. 2016, 9, 356–363. [Google Scholar] [CrossRef]
- Barbella, G.; Cocco, I.; Freri, E.; Marotta, G.; Visani, E.; Franceschetti, S.; Casazza, M. Transcutaneous vagal nerve stimulatio (t-VNS): An adjunctive treatment option for refractory epilepsy. Seizure 2018, 60, 115–119. [Google Scholar] [CrossRef]
- Lampros, M.; Vlachos, N.; Zigouris, A.; Voulgaris, S.; Alexiou, G.A. Transcutaneous vagus nerve stimulation (t-VNS) and epilepsy: A systematic review of the literature. Seizure 2021, 91, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Rong, P.; Liu, A.; Zhang, J.; Wang, Y.; He, W.; Yang, A.; Li, L.; Ben, H.; Li, L.; Liu, H. Transcutaneous vagus nerve stimulation for refractory epilepsy: A randomized controlled trial. Clin. Sci. 2014, CS20130518. [Google Scholar] [CrossRef] [PubMed]
- Gil-López, F.; Boget, T.; Manzanares, I.; Donaire, A.; Conde-Blanco, E.; Baillés, E.; Pintor, L.; Setoaín, X.; Bargalló, N.; Navarro, J. External trigeminal nerve stimulation for drug resistant epilepsy: A randomized controlled trial. Brain Stimul. 2020, 13, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Intersectoral Global Action Plan on Epilepsy and Other Neurological Disorders 2022–2031; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Keezer, M.R.; Sisodiya, S.M.; Sander, J.W. Comorbidities of epilepsy: Current concepts and future perspectives. Lancet. Neurol. 2016, 15, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Mula, M.; Coleman, H.; Wilson, S.J. Neuropsychiatric and Cognitive Comorbidities in Epilepsy. Continuum 2022, 28, 457–482. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Capelli, V.; Husain, M. Cognition and dementia in older patients with epilepsy. Brain 2018, 141, 1592–1608. [Google Scholar] [CrossRef]
- Holmes, G.L. Drug Treatment of Epilepsy Neuropsychiatric Comorbidities in Children. Paediatr. Drugs 2021, 23, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Fu, C.; Li, J.; Peng, S. Late-onset epilepsy and the risk of dementia: A systematic review and meta-analysis. Aging Clin. Exp. Res. 2022, 34, 1771–1779. [Google Scholar] [CrossRef]
- Do, P.T.; Chen, L.-Y.; Chan, L.; Hu, C.-J.; Chien, L.-N. Risk Factors for Postischemic Stroke Epilepsy in Young Adults: A Nationwide Population-Based Study in Taiwan. Front. Neurol. 2022, 13, 880661. [Google Scholar] [CrossRef]
- Beghi, E.; Beghi, M. Epilepsy, antiepileptic drugs and dementia. Curr. Opin. Neurol. 2020, 33, 191–197. [Google Scholar] [CrossRef]
- Strzelczyk, A.; Schubert-Bast, S. Psychobehavioural and Cognitive Adverse Events of Anti-Seizure Medications for the Treatment of Developmental and Epileptic Encephalopathies. CNS Drugs 2022, 36, 1079–1111. [Google Scholar] [CrossRef] [PubMed]
- Nanau, R.M.; Neuman, M.G. Adverse drug reactions induced by valproic acid. Clin. Biochem. 2013, 46, 1323–1338. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.-S.; Liu, I.-C.; Chiu, C.-H.; Huang, C.-J.; Wang, M.-Y. Effect of valproic acid on dementia onset in patients with bipolar disorder. J. Affect. Disord. 2016, 201, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Edinoff, A.N.; Nix, C.A.; Hollier, J.; Sagrera, C.E.; Delacroix, B.M.; Abubakar, T.; Cornett, E.M.; Kaye, A.M.; Kaye, A.D. Benzodiazepines: Uses, Dangers, and Clinical Considerations. Neurol. Int. 2021, 13, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Dagar, A.; Falcone, T. Psychiatric Comorbidities in Pediatric Epilepsy. Curr. Psychiatry Rep. 2020, 22, 77. [Google Scholar] [CrossRef] [PubMed]
- Verrotti, A.; Moavero, R.; Panzarino, G.; Di Paolantonio, C.; Rizzo, R.; Curatolo, P. The Challenge of Pharmacotherapy in Children and Adolescents with Epilepsy-ADHD Comorbidity. Clin. Drug Investig. 2018, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mula, M. Depression in epilepsy. Curr. Opin. Neurol. 2017, 30, 180–186. [Google Scholar] [CrossRef]
- Błaszczyk, B.; Czuczwar, S.J. Epilepsy coexisting with depression. Pharmacol. Rep. 2016, 68, 1084–1092. [Google Scholar] [CrossRef]
- Krishnan, V. Depression and Anxiety in the Epilepsies: From Bench to Bedside. Curr. Neurol. Neurosci. Rep. 2020, 20, 41. [Google Scholar] [CrossRef]
- Civan, A.B.; Ekici, A.; Havali, C.; Kiliç, N.; Bostanci, M. Evaluation of the risk factors for recurrence and the development of epilepsy in patients with febrile seizure. Arq. Neuro-Psiquiatria 2022, 80, 779–785. [Google Scholar] [CrossRef]
- Xue-Ping, W.; Hai-Jiao, W.; Li-Na, Z.; Xu, D.; Ling, L. Risk factors for drug-resistant epilepsy: A systematic review and meta-analysis. Medicine 2019, 98, e16402. [Google Scholar] [CrossRef] [PubMed]
- Lukmanji, S.; Manji, S.A.; Kadhim, S.; Sauro, K.M.; Wirrell, E.C.; Kwon, C.-S.; Jetté, N. The co-occurrence of epilepsy and autism: A systematic review. Epilepsy Behav. 2019, 98, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, X.; Sun, C.; Zou, M.; Chen, Y.; Huang, J.; Wu, L.; Chen, W.-X. Prevalence of epilepsy in autism spectrum disorders: A systematic review and meta-analysis. Autism 2022, 26, 33–50. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Arregui, T.; Llorente, J.; Giménez Minguez, P.; Tønnesen, J.; Peñagarikano, O. Neurobiological Mechanisms of Autism Spectrum Disorder and Epilepsy, Insights from Animal Models. Neuroscience 2020, 445, 69–82. [Google Scholar] [CrossRef]
- Lin, H.; Salech, F.; Lim, A.; Vogrin, S.; Duque, G. The effect of rapamycin and its analogues on age-related musculoskeletal diseases: A systematic review. Aging Clin. Exp. Res. 2022, 34, 2317–2333. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Ahn, S.; Min, S.K.; Ha, J.; Oh, G.T. Combined application of rapamycin and atorvastatin improves lipid metabolism in apolipoprotein E-deficient mice with chronic kidney disease. BMB Rep. 2021, 54, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Veroniki, A.A.; Rios, P.; Cogo, E.; Straus, S.E.; Finkelstein, Y.; Kealey, R.; Reynen, E.; Soobiah, C.; Thavorn, K.; Hutton, B.; et al. Comparative safety of antiepileptic drugs for neurological development in children exposed during pregnancy and breast feeding: A systematic review and network meta-analysis. BMJ Open 2017, 7, e017248. [Google Scholar] [CrossRef]
- Schachter, S.C. Anticonvulsant Agents: Principal Considerations on Effectiveness, Side Effects, and Interactions. In NeuroPsychopharmacotherapy; Riederer, P., Laux, G., Mulsant, B., Le, W., Nagatsu, T., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–8. [Google Scholar] [CrossRef]
- Sommerfeld-Klatta, K.; Zielińska-Psuja, B.; Karaźniewcz-Łada, M.; Główka, F.K. New Methods Used in Pharmacokinetics and Therapeutic Monitoring of the First and Newer Generations of Antiepileptic Drugs (AEDs). Molecules 2020, 25, 5083. [Google Scholar] [CrossRef]
- Lee, S.K. Old versus New: Why Do We Need New Antiepileptic Drugs? J. Epilepsy Res. 2014, 4, 39–44. [Google Scholar] [CrossRef]


{kind=link}
| NCT Number | Conditions | Interventions | Sex | Age | Phase |
|---|---|---|---|---|---|
| NCT02451696 | Epilepsy | Tuberous Sclerosis Complex | Focal Cortical Dysplasia | Everolimus | M/F | Child, adult | 2 |
| NCT02758626 | Epilepsy | Ataluren | Placebo | M/F | Child | 2 |
| NCT03940326 | Epilepsy, Idiopathic Generalized | Levetiracetam | Valproate | M/F | Child, adult | 4 |
| NCT02564952 | Epilepsy | GWP42003-P | Clobazam | M/F | Adult | 2 |
| NCT03650452 | Epilepsy | Dravet Syndrome | Lennox–Gastaut Syndrome | Tak-935 | Placebo | M/F | Child | 2 |
| NCT01777139 | Epilepsy | Retigabine Immediate Release | M/F | Adult | 3 |
| NCT03179891 | Epilepsy | Diazepam Buccal Film | M/F | Adult | 2 |
| NCT03283371 | Epilepsy, Focal Seizures, Partial Seizures | Natalizumab | Placebo | M/F | Adult | 2 |
| NCT01963208 | Drug Resistant Partial Onset Seizure | Ganaxolone | Placebo | M/F | Adult | 3 |
| NCT03478982 | Epilepsy | Staccato alprazolam | Placebo | M/F | Adult | 2 |
| NCT03222349 | Epilepsy | Diazepam Buccal Film | M/F | Child | 2 |
| NCT02682927 | Dravet Syndrome | Seizure Disorder | Zx008 (fenfluramine hydrochloride) | Placebo | M/F | Child, adult | 3 |
| NCT02224703 | Epilepsy | Dravet Syndrome | GWP42003-P | Placebo | M/F | Child, adult | 3 |
| NCT02565108 | Epilepsy | GWP42003-P | Clobazam | M/F | Adult | 2 |
| NCT02849626 | Partial-Onset or Primary Generalized Tonic–Clonic Seizures | Perampanel | M/F | Child | 3 |
| NCT02700412 | Epilepsy | Seizures | Epidiolex | M/F | Adult | 1 |
| NCT02724423 | Acute Repetitive Seizures | Nrl-1 | M/F | Child, adult | 1 |
| NCT02695537 | Epilepsy | Seizures | Epidiolex | M/F | Child, adult | 1 |
| NCT03405714 | Epilepsy | Brivaracetam | M/F | Child | 2 |
| NCT03373383 | Drug-Resistant Epilepsy | Focal-Onset Seizures | Padsevonil | Placebo | M/F | Adult | 2 |
| NCT01747915 | Generalized Tonic–Clonic Seizures | Pregabalin | Placebo | M/F | Child, adult | 3 |
| NCT02404168 | Epilepsy | Lamotrigine (Brand Lamictal) | Lamotrigine (Generic Teva) | M/F | Adult | 4 |
| NCT02721069 | Acute Repetitive Seizures | Breakthrough Seizures | NRL-1 | M/F | Child, adult | 3 |
| NCT01954121 | Epilepsy | Partial Seizures | Levetiracetam | Carbamazepine | M/F | Child, adult | 3 |
| NCT01832038 | Epilepsy | Partial-Onset Seizures | Lacosamide | M/F | Child, adult | 3 |
| NCT03428360 | Epilepsy | Diazepam Buccal Soluble Film | M/F | Child, adult | 3 |
| NCT02036853 | Glucose Transporter Type-1 Deficiency Syndrome (Glut1 DS) | Triheptanoin | M/F | Child, adult | 2 |
| NCT01964560 | Epilepsy | Lacosamide | M/F | Child | 3 |
| NCT02224560 | Epilepsy | Lennox–Gastaut Syndrome | GWP42003-P | Placebo | M/F | Child, adult | 3 |
| NCT02224573 | Epilepsy | Dravet Syndrome | Lennox–Gastaut Syndrome | GWP42003-P | M/F | Child, adult | 3 |
| NCT03021018 | Epilepsy | Brivaracetam | Lorazepam | M/F | Adult | 2 |
| NCT01999777 | Epilepsy | USL261 | Placebo | M/F | Child, adult | 3 |
| NCT01713946 | Tuberous Sclerosis Complex-Associated Refractory Seizures | RAD001 | Placebo | Antiepileptic drug (1 to 3 only) | Open Label RAD001 | M/F | Child, adult | 3 |
| NCT02564029 | Reflex Epilepsy, Photosensitive | PF-06372865 | Placebo | Lorazepam | M/F | Adult | 2 |
| NCT03116828 | Epilepsy with Partial Onset Seizures | Eslicarbazepine Acetate (first add-on) | Eslicarbazepine Acetate (late add-on) | M/F | Adult | 4 |
| NCT02100644 | Epilepsy | Lamotrigine Tablets | F | Child, adult | 4 |
| NCT02495844 | Highly Drug-Resistant Focal Epilepsy | UCB0942 | Placebo | M/F | Adult | 2 |
| NCT01866111 | Partial Epilepsy | Ykp3089 | Placebo | M/F | Adult | 2 |
| NCT02926898 | Dravet Syndrome | Zx008 (fenfluramine hydrochloride) | Matching Placebo | M/F | Child, adult | 3 |
| NCT02351115 | Epilepsy | Placebo | Inhaled Alprazolam | Inhaled Alprazolam | Inhaled Alprazolam | Placebo | M/F | Adult | 2 |
| NCT03953820 | Epilepsy | Diazepam Buccal Film | Diastat® Rectal Gel | M/F | Adult | 1|2 |
| NCT02072824 | Partial Onset Seizures | Pregabalin dose level 1 | Pregabalin dose level 2 | Placebo | M/F | Child | 3 |
| NCT04882540 | Healthy Participants | Brivaracetam | M/F | Adult | 1 |
| NCT02726074 | Epilepsy | Perampanel | M/F | Child, adult | 4 |
| Side Effects | Drug | |
|---|---|---|
| Suicidal behavior and ideation | Brivaracetam | Lamotrigine |
| Cannabidiol Oral Solution | Levetiracetam-XR | |
| Carbamazepine | Midazolam Nasal | |
| Carbamazepine-XR | Oxcarbazepine | |
| Cenobamate | Perampanel | |
| Clobazam | Phenytoin | |
| Clonazepam | Pregabalin | |
| Diazepam Nasal | Primidone | |
| Divalproex Sodium | Rufinamide | |
| Eslicarbazepine Acetate | Stiripentol | |
| Ethosuximide | Topiramate | |
| Felbamate | Topiramate XR | |
| Fenfluramine | Valproic Acid | |
| Gabapentin | Fenfuramine | |
| Lacosamide | Zonisamide | |
| Psychiatric adverse reaction | Brivaracetam | Perampanel |
| Fenfluramine | Phenobarbital | |
| Gabapentin | Pregabalin | |
| Levetiracetam | Tiagabine Hydrochloride | |
| Levetiracetam-XR | Topiramate | |
| Lorazepam | Topiramate XR | |
| Midazolam Nasal | Zonisamide | |
| Oxcarbazepine | ||
| Neurological Adverse Reactions | Brivaracetam | Lorazepam |
| Cannabidiol Oral Solution | Midazolam Nasal | |
| Cenobamate | Oxcarbazepine | |
| Clobazam | Perampanel | |
| Clonazepam | Phenobarbital | |
| Diazepam Nasal | Pregabalin | |
| Diazepam Rectal | Primidone | |
| Divalproex Sodium | Rufinamide | |
| Divalproex Sodium-ER | Stiripentol | |
| Eslicarbazepine Acetate | Tiagabine Hydrochloride | |
| Felbamate | Topiramate | |
| Fenfluramine | Topiramate XR | |
| Gabapentin | Valproic Acid | |
| Lacosamide | Fenfuramine | |
| Levetiracetam | Zonisamide | |
| Levetiracetam-XR | ||
| Hypersensitivity | Brivaracetam | Lacosamide |
| Cannabidiol oral solution | Lamotrigine | |
| Carbamazepine | Oxcarbazepine | |
| Carbamazepine-XR | Phenobarbital | |
| Cenobamate | Phenytoin | |
| Eslicarbazepine Acetate | Pregabalin | |
| Gabapentin | ||
| Withdrawal of AEDs | Brivaracetam | Lorazepam |
| Cannabidiol oral solution | Midazolam Nasal | |
| Cenobamate | Oxcarbazepine | |
| Clobazam | Perampanel | |
| Clonazepam | Phenobarbital | |
| Diazepam Nasal | Phenytoin | |
| Diazepam Rectal | Rufinamide | |
| Eslicarbazepine Acetate | Stiripentol | |
| Fenfluramine | Tiagabine Hydrochloride | |
| Gabapentin | Topiramate | |
| Lacosamide | Topiramate XR | |
| Lamotrigine | Fenfuramine | |
| Levetiracetam | Zonisamide | |
| Levetiracetam-XR | ||
| Hepatotoxicity | Cannabidiol Oral Solution | Phenytoin |
| Divalproex Sodium | Valproic Acid | |
| Divalproex Sodium-ER | ||
| Serious Dermatologic Reactions | Carbamazepine | Levetiracetam-XR |
| Carbamazepine-XR | Oxcarbazepine | |
| Clobazam | Phenobarbital | |
| Eslicarbazepine Acetate | Phenytoin | |
| Ethosuximide | Topiramate | |
| Lamotrigine | Zonisamide | |
| Levetiracetam | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boleti, A.P.d.A.; Cardoso, P.H.d.O.; Frihling, B.E.F.; de Moraes, L.F.R.N.; Nunes, E.A.C.; Mukoyama, L.T.H.; Nunes, E.A.C.; Carvalho, C.M.E.; Macedo, M.L.R.; Migliolo, L. Pathophysiology to Risk Factor and Therapeutics to Treatment Strategies on Epilepsy. Brain Sci. 2024, 14, 71. https://doi.org/10.3390/brainsci14010071
Boleti APdA, Cardoso PHdO, Frihling BEF, de Moraes LFRN, Nunes EAC, Mukoyama LTH, Nunes EAC, Carvalho CME, Macedo MLR, Migliolo L. Pathophysiology to Risk Factor and Therapeutics to Treatment Strategies on Epilepsy. Brain Sciences. 2024; 14(1):71. https://doi.org/10.3390/brainsci14010071
Chicago/Turabian StyleBoleti, Ana Paula de Araújo, Pedro Henrique de Oliveira Cardoso, Breno Emanuel Farias Frihling, Luiz Filipe Ramalho Nunes de Moraes, Ellynes Amancio Correia Nunes, Lincoln Takashi Hota Mukoyama, Ellydberto Amancio Correia Nunes, Cristiano Marcelo Espinola Carvalho, Maria Lígia Rodrigues Macedo, and Ludovico Migliolo. 2024. "Pathophysiology to Risk Factor and Therapeutics to Treatment Strategies on Epilepsy" Brain Sciences 14, no. 1: 71. https://doi.org/10.3390/brainsci14010071