WO2013008095A1 - Novel pyrrolo pyrimidine derivatives - Google Patents
Novel pyrrolo pyrimidine derivatives Download PDFInfo
- Publication number
- WO2013008095A1 WO2013008095A1 PCT/IB2012/001699 IB2012001699W WO2013008095A1 WO 2013008095 A1 WO2013008095 A1 WO 2013008095A1 IB 2012001699 W IB2012001699 W IB 2012001699W WO 2013008095 A1 WO2013008095 A1 WO 2013008095A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- phenyl
- fluoro
- pyrrolo
- pyrimidin
- Prior art date
Links
- 0 CC1(C)OB(c2cc(*)c(*)c(N)c2*)OC1(C)C Chemical compound CC1(C)OB(c2cc(*)c(*)c(N)c2*)OC1(C)C 0.000 description 3
- PWOGVDHSRZNLTQ-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1C(Nc1cccc(-c2ncnc3c2cc(C2=CCOCC2)[nH]3)c1C)=O Chemical compound CC(C)(C)c(cc1)ccc1C(Nc1cccc(-c2ncnc3c2cc(C2=CCOCC2)[nH]3)c1C)=O PWOGVDHSRZNLTQ-UHFFFAOYSA-N 0.000 description 2
- IZMQTBFHDQBFNO-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CC=C1c([nH]c1ncn2)cc1c2Cl)=O Chemical compound CC(C)(C)OC(N(CC1)CC=C1c([nH]c1ncn2)cc1c2Cl)=O IZMQTBFHDQBFNO-UHFFFAOYSA-N 0.000 description 1
- CWCDRMMXNVSKMN-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1C(Nc1c(C)c(-c2ncnc3c2cc(C(CC2)=CCS2=O)[nH]3)cc(F)c1)=O Chemical compound CC(C)(C)c(cc1)ccc1C(Nc1c(C)c(-c2ncnc3c2cc(C(CC2)=CCS2=O)[nH]3)cc(F)c1)=O CWCDRMMXNVSKMN-UHFFFAOYSA-N 0.000 description 1
- OMMQPNOVPRBWPC-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1C(Nc1c(C)c(B2OC(C)(C)C(C)(C)O2)cc(F)c1)=O Chemical compound CC(C)(C)c(cc1)ccc1C(Nc1c(C)c(B2OC(C)(C)C(C)(C)O2)cc(F)c1)=O OMMQPNOVPRBWPC-UHFFFAOYSA-N 0.000 description 1
- ATTNKONWSRSBKY-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1C(Nc1c(C)c(B2OC(C)(C)C(C)(C)O2)ccc1)=O Chemical compound CC(C)(C)c(cc1)ccc1C(Nc1c(C)c(B2OC(C)(C)C(C)(C)O2)ccc1)=O ATTNKONWSRSBKY-UHFFFAOYSA-N 0.000 description 1
- XITCTVRXMGEAJJ-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1C(Nc1cccc(-c2ncnc3c2cc(C(CC2)=CCC2=O)[nH]3)c1CO)=O Chemical compound CC(C)(C)c(cc1)ccc1C(Nc1cccc(-c2ncnc3c2cc(C(CC2)=CCC2=O)[nH]3)c1CO)=O XITCTVRXMGEAJJ-UHFFFAOYSA-N 0.000 description 1
- UJAKODWTNVLXEA-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1C(Nc1cccc(-c2ncnc3c2cc(C(CC2)=CCN2C(OC(C)(C)C)=O)[nH]3)c1COCC(C)(C)[S-](c1ccccc1)c1ccccc1)=O Chemical compound CC(C)(C)c(cc1)ccc1C(Nc1cccc(-c2ncnc3c2cc(C(CC2)=CCN2C(OC(C)(C)C)=O)[nH]3)c1COCC(C)(C)[S-](c1ccccc1)c1ccccc1)=O UJAKODWTNVLXEA-UHFFFAOYSA-N 0.000 description 1
- PFBLKWQTZFUGHL-UHFFFAOYSA-N CC(C)(COC)c(cc1)ccc1C(Nc1c(C)c(-c2ncnc3c2cc(C(CC2)=CCN2C(N(C)C)=O)[nH]3)cc(F)c1)=O Chemical compound CC(C)(COC)c(cc1)ccc1C(Nc1c(C)c(-c2ncnc3c2cc(C(CC2)=CCN2C(N(C)C)=O)[nH]3)cc(F)c1)=O PFBLKWQTZFUGHL-UHFFFAOYSA-N 0.000 description 1
- YXDNGVVRHSAJIY-UHFFFAOYSA-N CC(C)(c(cc1)cc(CCN2c3c(COC(C)=O)c(B4OC(C)(C)C(C)(C)O4)ccc3)c1C2=O)O Chemical compound CC(C)(c(cc1)cc(CCN2c3c(COC(C)=O)c(B4OC(C)(C)C(C)(C)O4)ccc3)c1C2=O)O YXDNGVVRHSAJIY-UHFFFAOYSA-N 0.000 description 1
- LOSQXNNWLNWKPQ-UHFFFAOYSA-N CC(C)(c(cc1CCN2c3c(CO)c(Br)ccc3)ccc1C2=O)O Chemical compound CC(C)(c(cc1CCN2c3c(CO)c(Br)ccc3)ccc1C2=O)O LOSQXNNWLNWKPQ-UHFFFAOYSA-N 0.000 description 1
- BXZGCFXOZWMQQC-UHFFFAOYSA-N CC(C)(c1ccc(C(Nc2cccc(-c3ncnc4c3cc(C3=CCOCC3)[nH]4)c2CO)=O)c(F)c1)O Chemical compound CC(C)(c1ccc(C(Nc2cccc(-c3ncnc4c3cc(C3=CCOCC3)[nH]4)c2CO)=O)c(F)c1)O BXZGCFXOZWMQQC-UHFFFAOYSA-N 0.000 description 1
- UJVGRWVBHNSHNC-UHFFFAOYSA-N Cc(c(-c1ncnc2c1cc(C(CC1)=CCN1C(CO)=O)[nH]2)cc(F)c1)c1NC(c(cc1)ccc1S(F)(F)(F)(F)F)=O Chemical compound Cc(c(-c1ncnc2c1cc(C(CC1)=CCN1C(CO)=O)[nH]2)cc(F)c1)c1NC(c(cc1)ccc1S(F)(F)(F)(F)F)=O UJVGRWVBHNSHNC-UHFFFAOYSA-N 0.000 description 1
- LEFWFEAAZYOYRL-UHFFFAOYSA-N Cc(c(NC(N(C1)CC1OC(C(F)(F)F)C(F)(F)F)=O)cc(F)c1)c1-c1c(cc(C(CC2)=CCN2C(N(C)C)=O)[nH]2)c2ncn1 Chemical compound Cc(c(NC(N(C1)CC1OC(C(F)(F)F)C(F)(F)F)=O)cc(F)c1)c1-c1c(cc(C(CC2)=CCN2C(N(C)C)=O)[nH]2)c2ncn1 LEFWFEAAZYOYRL-UHFFFAOYSA-N 0.000 description 1
- CQYJSPQAOHYHKO-UHFFFAOYSA-N Cc(c(NC(N1Cc2cc(F)ccc2C1)=O)cc(F)c1)c1-c1ncnc2c1cc(C(CC1)=CCN1C(N(C)C)=O)[nH]2 Chemical compound Cc(c(NC(N1Cc2cc(F)ccc2C1)=O)cc(F)c1)c1-c1ncnc2c1cc(C(CC1)=CCN1C(N(C)C)=O)[nH]2 CQYJSPQAOHYHKO-UHFFFAOYSA-N 0.000 description 1
- JWXICKMBHDHSJV-UHFFFAOYSA-N Cc(c(NC(c(cc1)ccc1N(C)C)=O)cc(F)c1)c1-c1ncnc2c1cc(C(CC1)=CCN1C(N(C)C)=O)[nH]2 Chemical compound Cc(c(NC(c(cc1)ccc1N(C)C)=O)cc(F)c1)c1-c1ncnc2c1cc(C(CC1)=CCN1C(N(C)C)=O)[nH]2 JWXICKMBHDHSJV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- the present invention describes new pyrrolo pyrimidine derivatives that are good drug candidates.
- the compounds of the present invention may generally exhibit a selective inhibition of Bruton's tyrosine kinase (Btk).
- Btk-deficient mice are protected in standard preclinical models for rheumatoid arthritis (Jansson and Holmdahl, 1993), systemic lupus erythematosus (Steinberg, B.J. et al., J. Clin. Invest, 70, 587-597, 1982), as well as allergic disease and anaphylaxis (Hata.D. et al., J. Exp. Med. 187, 1235-1247, 1998).
- many cancers and lymphomas express Btk and appear to be dependent on Btk function (Davis, R.E. et al., Nature, 463, 88-92, 2010).
- inhibition of Btk activity may be useful in the treatment of immune disorders such as rheumatoid arthritis, systemic lupus erythematosus, allergic diseases, anaphylaxis and inflammatory conditions.
- inhibition of Btk may be useful in the treatment of cancers of haematopoietic origin including chronic myelogenous leukemia, myeloid leukemia, non-Hodgkin lymphoma and other B cell lymphomas.
- the compounds of the present invention may therefore potentially be useful in the treatment of a wide range of disorders, particularly Btk-related diseases or disorders, and may for example be useful in the treatment of autoimmune disorders, inflammatory diseases, allergic diseases, airway diseases, such as asthma and chronic obstructive pulmonary disease (COPD), transplant rejection, or cancers e.g. of hematopoietic origin or solid tumors. More particularly, the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof;
- R1 is hydrogen, CrC 6 alkyi optionally substituted by hydroxy
- R2 is hydrogen or halogen
- R3 is hydrogen or halogen
- R4 is hydrogen
- R5 is phenyl optionally substituted by halogen; SF 5 ; NR6R7; hydroxy; C C 6 alkoxy; d- C 6 alkenyl; CrC 6 alkyi carbonyl; CrC 6 alkyi optionally substituted by hydroxy, halogen, or Ci-C 6 alkoxy; or C 3 -C 6 cycloalkyi optionally substituted by halogen, hydroxy, or C C 6 alkyi optionally substituted by halogen; or
- R5 is a 4 - 14 membered mono- or bicyclic heterocyclyl or heteroaryl ring system comprising 1 , 2 or 3 heteroatoms selected from N, S and O that ring being optionally substituted by halogen; hydroxy; CrC 6 alkoxy optionally substituted by hydroxy or halogen; or CrC 6 alkyi optionally substituted by hydroxy or halogen;
- R4 and R5 together with the atoms to which they are bound form a piperidone ring, optionally comprising an annulated phenyl ring, any such ring being optionally substituted by C C 6 alkyi, C C 6 alkoxy, or C 3 -C 6 cycloalkyi each of which substitution member may optionally be substituted by halogen or hydroxy;
- R6 and R7 are independently selected from hydrogen or CrC 6 alkyi
- R6 and R7 together with the nitrogen atom to which they are bound form a 4 - 8 membered saturated azacycloalkane ring, optionally substituted by halogen, hydroxy or
- X is O, S(0) n wherein n is 0, 1 or 2, or wherein q is 2 or 3, and R10 is absent; or X is CH or N; and R10 is hydrogen, hydroxy, -NR6R7, -CO-R1 1 , -S(0) p -R12 wherein p is 1 or 2,
- R 1 1 is Ci-C 6 alkyl optionally substituted by hydroxy, cyano, halogen, carboxy or CrC 6 alkoxy carbonyloxy; or NR6R7; and
- R12 is Ci-C 6 alkyl or NR6R7.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is hydrogen, methyl or
- R2 and R3 are independently hydrogen or fluoro
- R4 is hydrogen
- R5 is phenyl substituted by halogen
- CrC 6 alkoxy or CrC 6 alkyl optionally substituted by halogen or hydroxy
- C 3 -C 6 cycloalkyi optionally substituted by halogen, hydroxy, C C 6 substituted by halogen
- X is O, S(0) n wherein n is 0, 1 or 2, or wherein q is 2 or 3, and R10 is absent;
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is hydrogen, methyl or
- R2 and R3 are independently hydrogen or fluoro
- R4 together with R5 is a 3,4-dihydro-2H-isoquinolin-1-one optionally substituted by C 3 -C 6 cycloalkyi or C C 6 substituted by hydroxy
- X is O, S(0) n wherein n is 0, 1 or 2, or wherein q is 2 or 3, and R10 is absent; and the remaining variables are as defined above.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is hydrogen, methyl or
- R2 and R3 are independently hydrogen or fluoro
- R4 is hydrogen
- R5 is phenyl substituted by halogen
- CrC 6 alkoxy CrC 6 alkyl optionally substituted by halogen or hydroxy
- C 3 -C 6 cycloalkyi optionally substituted by halogen, hydroxy, or d- C 6 alkyl optionally substituted by halogen
- X stands for O and R10 is absent
- X stands for N
- R10 is hydrogen or -CO-R1 1 , and the remaining variables are as defined above.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is hydrogen, methyl or
- R2 and R3 are independently hydrogen or fluoro
- R4 is hydrogen
- R5 is phenyl substituted by CrC 6 alkoxy; CrC 6 alkyl optionally substituted by halogen or hydroxy; or C 3 -C 6 cycloalkyl optionally substituted by halogen, hydroxy, or CrC 6 alkyl optionally substituted by halogen
- X stands for N
- R10 is hydrogen or -CO-R1 1
- R1 1 stands for NR6R7 wherein R6 and R7 are independently hydrogen or methyl; and the remaining variables are as defined above.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is hydrogen, methyl or
- R2 and R3 are independently hydrogen or fluoro
- R4 is hydrogen
- R5 is azetidine optionally substituted by CrC 6 alkoxy
- X stands for N
- R10 is hydrogen or -CO-R1 1 , and the remaining variables are as defined above.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is hydrogen, methyl or
- R2 and R3 are independently hydrogen or fluoro
- R4 together with R5 is a 3,4-dihydro-2H-isoquinolin-1-one optionally substituted by C 3 -C 6 cycloalkyl or C C 6 alkyl optionally substituted by hydroxy
- X stands for N
- R10 is hydrogen or -CO-R1 1 , and the remaining variables are as defined above.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is hydrogen, methyl or
- R2 and R3 are independently hydrogen or fluoro
- R4 together with R5 is a 3,4-dihydro-2H-isoquinolin-1-one substituted by C 3 -C 6 cycloalkyl or C C 6 alkyl optionally substituted by hydroxy in the 6-position of said isoquinolin-ring
- X stands for N
- R10 is hydrogen or -CO-R1 1 , and the remaining variables are as defined above.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is hydrogen, methyl or
- R2 and R3 are independently hydrogen or fluoro
- R4 is hydrogen
- R5 is azetidine optionally substituted by CrC 6 alkoxy
- X stands for O, and the remaining variables are as defined above.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is hydrogen, methyl or
- R2 and R3 are independently hydrogen or fluoro
- R4 together with R5 is a 3,4-dihydro-2H-isoquinolin-1-one optionally substituted by C 3 -C 6 cycloalkyi or C C 6 alkyl optionally substituted by hydroxy
- X stands for O, and the remaining variables are as defined above.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is hydrogen, methyl or
- R2 and R3 are independently hydrogen or fluoro
- R4 together with R5 is a 3,4-dihydro-2H-isoquinolin-1-one substituted by C 3 -C 6 cycloalkyi or C C 6 alkyl optionally substituted by hydroxy in the 6-position of said isoquinolin-ring
- X stands for O, and the remaining variables are as defined above.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is hydrogen, methyl or
- R2 and R3 are independently from each other selected from hydrogen and halogen
- R4 is hydrogen
- R5 is phenyl substituted one or more times by halogen
- X stands for O or S
- R10 is absent.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof wherein R1 is methyl or hydroxymethyl , R2 and R3 are independently from each other selected from hydrogen and halogen, R4 is hydrogen, R5 is phenyl substituted one or more times by halogen, C 3 -C 6 cycloalkyi, or Ci-C 6 alkyl optionally substituted by hydroxy, X stands for N, R10 is hydrogen or -CO-R1 1 , and R1 1 is NR6R7 wherein R6 and R7 are independently selected from C Ce-alkyl.
- R1 is methyl or hydroxymethyl
- R2 and R3 are independently from each other selected from hydrogen and halogen
- R4 is hydrogen
- R5 is phenyl substituted one or more times by halogen, C 3 -C 6 cycloalkyi, or Ci-C 6 alkyl optionally substituted by hydroxy
- X stands for N
- R10 is hydrogen or -CO-R
- R1 is hydrogen, methyl or hydroxymethyl
- R1 is methyl or hydroxymethyl
- R2 and R3 are independently hydrogen or fluoro
- R1 is methyl or hydroxymethyl and R2 and R3 are independently hydrogen or fluoro;
- R4 is hydrogen
- R4 together with R5 is a 3,4-dihydro-2H-isoquinolin-1-one optionally substituted by C 3 -C 6 cycloalkyi or CrC 6 alkyl optionally substituted by hydroxy;
- R4 together with R5 is a 3,4-dihydro-2H-isoquinolin-1-one optionally substituted by C 3 -C 6 cycloalkyi or CrC 6 alkyl optionally substituted by hydroxy in the 6- position of the 3,4-dihydro-2H-isoquinolin-1-one ring;
- R5 is phenyl optionally substituted by -NR6R7, halogen; CrC 6 alkoxy, CrC 6 alkenyl, C 3 -C 6 cycloalkyi, or CrC 6 alkyl optionally substituted by halogen or hydroxy;
- R5 is phenyl substituted by -NR6R7, halogen, CrC 6 alkoxy or C C 6 alkyl optionally substituted by halogen or hydroxy;
- R5 is phenyl substituted by halogen, CrC 6 alkoxy, C 3 -C 6 cycloalkyi or C C 6 alkyl optionally substituted by fluoro or hydroxy;
- R5 is a 4-, 5-, 6-, or 7-membered monocyclic heterocycle, or a 7-, 8-, 9-, 10-, 1 1-, or 12-membered bicyclic heterocycle comprising 1 , 2 or 3 heteroatoms selected from N, S and O that ring being optionally substituted by halogen; hydroxy; CrC 6 alkoxy optionally substituted by hydroxy or halogen; or C C 6 alkyl optionally substituted by hydroxy or halogen;
- R5 is a 4-, 5-, 6-, or 7-membered monocyclic heterocycle comprising 1 or 2 heteroatoms selected from N, S and O that ring being optionally substituted by halogen; hydroxy; CrC 6 alkoxy optionally substituted by hydroxy or halogen; or Ci-C 6 alkyl optionally substituted by hydroxy or halogen;
- R5 is azetidine substituted by C C 6 alkoxy or C C 6 alkyl; 14.
- X is O and R10 is absent or is N and R10 is H or CO-R1 1 ;
- X is O and R10 is absent or is N and R10 is CO-R1 1 ;
- X is N and R10 is CO-R11 ;
- R11 is NR6R7 and R6 and R7 are independently selected from hydrogen or d- C 6 alkyl;
- R11 is NR6R7 and R6 and R7 are independently selected from d-C 6 alkyl;
- R11 is NR6R7 and R6 and R7 are independently selected from C C 3 alkyl;
- R11 is NR6R7 and R6 and R7 are methyl.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use as a medicament.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in the treatment of a disease or disorder mediated by Btk.
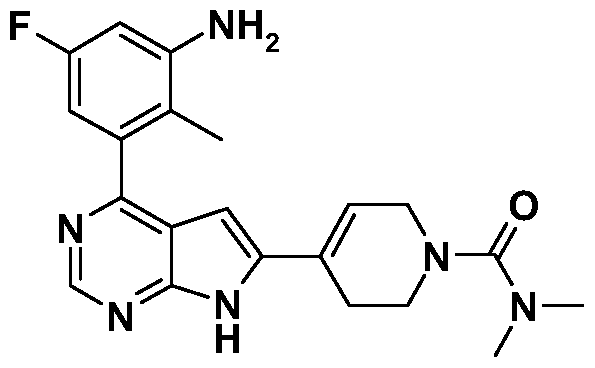
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof, which is selected from:
- alkyl refers to a fully saturated branched or unbranched hydrocarbon moiety having up to 20 carbon atoms. Unless otherwise provided, alkyl refers to hydrocarbon moieties having 1 to 16 carbon atoms, 1 to 10 carbon atoms, 1 to 7 carbon atoms, or 1 to 4 carbon atoms.
- alkyl include, but are not limited to, methyl, ethyl, n-propyl, / ' so-propyl, n-butyl, sec-butyl, / ' so-butyl, tert- butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, 3-methylhexyl, 2,2- dimethylpentyl, 2,3- dimethylpentyl, n-heptyl, n-octyl, n-nonyl, n-decyl and the like.
- alkenyl refers to an unsaturated branched or unbranched hydrocarbon moiety having 2 to 20 carbon atoms. It comprises 2 to 20 carbon atoms, Unless otherwise provided, alkenyl refers to moieties having 2 to 16 carbon atoms, 2 to 10 carbon atoms, 2 to 7 carbon atoms, or 2 to 4 carbon atoms.
- alkenyl include, but are not limited to, ethenyl, n-propenyl, / ' so-propenyl, n- butenyl, sec-butenyl, / ' so-butenyl, fert-butenyl, n-pentenyl, isopentenyl, neopentenyl, n- hexenyl, 3-methylhexenyl, 2,2- dimethylpentenyl, 2,3-dimethylpentenyl, n-heptenyl, n- octenyl, n-nonenyl, n-decenyl and the like.
- alkoxy refers to alkyl-O-, wherein alkyl is defined herein above.
- Representative examples of alkoxy include, but are not limited to, methoxy, ethoxy, propoxy, 2-propoxy, butoxy, fert-butoxy, pentyloxy, hexyloxy, cyclopropyloxy-, cyclohexyloxy- and the like.
- alkoxy groups typically have about 1-7, more preferably about 1-4 carbons.
- cycloalkyl refers to saturated or unsaturated monocyclic, bicyclic or tricyclic hydrocarbon groups of 3-12 carbon atoms. Unless otherwise provided, cycloalkyl refers to cyclic hydrocarbon groups having between 3 and 9 ring carbon atoms or between 3 and 7 ring carbon atoms. Exemplary monocyclic hydrocarbon groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl and cyclohexenyl and the like.
- Exemplary bicyclic hydrocarbon groups include bornyl, indyl, hexahydroindyl, tetrahydronaphthyl, decahydronaphthyl, bicyclo[2.1.1]hexyl, bicyclo[2.2.1]heptyl, bicyclo[2.2.1]heptenyl, 6,6- dimethylbicyclo[3.1.1]heptyl, 2,6,6-trimethylbicyclo[3.1.1]heptyl, bicyclo[2.2.2]octyl and the like.
- Exemplary tricyclic hydrocarbon groups include adamantyl and the like.
- azacycloalkane refers to saturated or unsaturated monocyclic, bicyclic or tricyclic hydrocarbon groups of 3-12 carbon atoms as defined for “cycloalkyl", wherein one carbon atom is replaced by a nitrogen atom.
- azacycloalkyi refers to cyclic aza-hydrocarbon groups having between 2 and 9 ring carbon atoms and one nitrogen atom or between 2 and 7 ring carbon atoms and one nitrogen atom.
- Exemplary monocyclic aza-hydrocarbon groups include, but are not limited to, aziridinyl, azetidinly, pyrollidinyl, piperidinyl, azepanyl, dihydroazepinyl and the like.
- halogen refers to fluoro, chloro, bromo, and iodo.
- heterocyclic may refer to a saturated or unsaturated non-aromatic ring or ring system, e.g., which is a 4-, 5-, 6-, or 7-membered monocyclic, 7-, 8-, 9-, 10-, 1 1-, or 12-membered bicyclic or 10-, 1 1-, 12-, 13-, 14- or 15-membered tricyclic ring system and contains at least one heteroatom selected from O, S and N, where the N and S can also optionally be oxidized to various oxidation states.
- the heterocyclic group can be attached at a heteroatom or a carbon atom.
- the heterocyclyl may include fused or bridged rings as well as spirocyclic rings.
- heterocycles include azetidine, tetrahydrofuran (THF), dihydrofuran, 1 , 4- dioxane, morpholine, 1 ,4-dithiane, piperazine, piperidine, 1 ,3-dioxolane, imidazolidine, imidazoline, pyrroline, pyrrolidine, tetrahydropyran, dihydropyran, oxathiolane, dithiolane, 1 ,3-dioxane, 1 ,3-dithiane, oxathiane, thiomorpholine, and the like.
- aryloxy refers to both an -O-aryl and an -O-heteroaryl group, wherein aryl and heteroaryl are defined herein.
- heteroaryl refers to a 5-14 membered monocyclic- or bicyclic- or tricyclic-aromatic ring system, having 1 to 8 heteroatoms selected from N, O or S.
- the heteroaryl is a 5-10 membered ring system (e.g., 5-7 membered monocycle or an 8-10 memberred bicycle) or a 5-7 membered ring system.
- Typical heteroaryl groups include 2- or 3-thienyl, 2- or 3-furyl, 2- or 3-pyrrolyl, 2-, 4-, or 5- imidazolyl, 3-, 4-, or 5- pyrazolyl, 2-, 4-, or 5-thiazolyl, 3-, 4-, or 5-isothiazolyl, 2-, 4-, or 5- oxazolyl, 3-, 4-, or 5-isoxazolyl, 3- or 5-1 ,2,4-triazolyl, 4- or 5-1 ,2, 3-triazolyl, tetrazolyl, 2- , 3-, or 4-pyridyl, 3- or 4-pyridazinyl, 3-, 4-, or 5-pyrazinyl, 2-pyrazinyl, and 2-, 4-, or 5- pyrimidinyl.
- heteroaryl also refers to a group in which a heteroaromatic ring is fused to one or more aryl, cycloaliphatic, or heterocyclyl rings, where the radical or point of attachment is on the heteroaromatic ring.
- Nonlimiting examples include 1-, 2-, 3-, 5-, 6-, 7-, or 8- indolizinyl, 1-, 3-, 4-, 5-, 6-, or 7-isoindolyl, 2-, 3-, 4-, 5-, 6-, or 7-indolyl, 2-, 3- , 4-, 5-, 6-, or 7-indazolyl, 2-, 4-, 5-, 6-, 7-, or 8- purinyl, 1-, 2-, 3-, 4-, 6-, 7-, 8-, or 9- quinolizinyl, 2-, 3-, 4-, 5-, 6-, 7-, or 8-quinoliyl, 1-, 3-, 4-, 5-, 6-, 7-, or 8-isoquinoliyl, 1-, 4-, 5-, 6-, 7-, or 8-phthalazinyl, 2-, 3-, 4-, 5-, or 6-naphthyridinyl, 2-, 3-, 4-, 5-, or 6-naphthyridinyl, 2-, 3- , 5-, 6-, 7-, or 8
- Typical fused heteroary groups include, but are not limited to 2-, 3-, 4-, 5-, 6-, 7-, or 8-quinolinyl, 1-, 3-, 4-, 5-, 6-, 7-, or 8-isoquinolinyl, 2-, 3-, 4-, 5-, 6-, or 7-indolyl, 2-, 3-, 4-, 5-, 6-, or 7-benzo[b]thienyl, 2-, 4-, 5- , 6-, or 7- benzoxazolyl, 2-, 4-, 5-, 6-, or 7-benzimidazolyl, and 2-, 4-, 5-, 6-, or 7-benzothiazolyl.
- salt refers to an acid addition or base addition salt of a compound of the invention.
- Salts include in particular “pharmaceutical acceptable salts”.
- pharmaceutically acceptable salts refers to salts that retain the biological effectiveness and properties of the compounds of this invention and, which typically are not biologically or otherwise undesirable.
- the compounds of the present invention are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto.
- Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids, e.g., acetate, aspartate, benzoate, besylate, bromide/hydrobromide, bicarbonate/carbonate, bisulfate/sulfate, camphorsulfonate, chloride/hydrochloride, chlortheophyllonate, citrate, ethandisulfonate, fumarate, gluceptate, gluconate, glucuronate, hippurate, hydroiodide/iodide, isethionate, lactate, lactobionate, laurylsulfate, malate, maleate, malonate, mandelate, mesylate, methylsulphate, naphthoate, napsylate, nicotinate, nitrate, octadecanoate, oleate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen
- Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
- Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, sulfosalicylic acid, and the like.
- Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
- Inorganic bases from which salts can be derived include, for example, ammonium salts and metals from columns I to XII of the periodic table.
- the salts are derived from sodium, potassium, ammonium, calcium, magnesium, iron, silver, zinc, and copper; particularly suitable salts include ammonium, potassium, sodium, calcium and magnesium salts.
- Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like.
- Certain organic amines include isopropylamine, benzathine, cholinate, diethanolamine, diethylamine, lysine, meglumine, piperazine and tromethamine.
- the pharmaceutically acceptable salts of the present invention can be synthesized from a basic or acidic moiety, by conventional chemical methods.
- such salts can be prepared by reacting free acid forms of these compounds with a stoichiometric amount of the appropriate base (such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like), or by reacting free base forms of these compounds with a stoichiometric amount of the appropriate acid.
- a stoichiometric amount of the appropriate base such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like
- Such reactions are typically carried out in water or in an organic solvent, or in a mixture of the two.
- use of non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile is desirable, where practicable.
- any formula given herein is also intended to represent unlabeled forms as well as isotopically labeled forms of the compounds.
- Isotopically labeled compounds have structures depicted by the formulas given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 18 F 31 P, 32 P, 35 S, 36 CI, 125 l respectively.
- the invention includes various isotopically labeled compounds as defined herein, for example those into which radioactive isotopes, such as 3 H and 14 C, or those into which non-radioactive isotopes, such as 2 H and 13 C are present.
- isotopically labelled compounds are useful in metabolic studies (with 14 C), reaction kinetic studies (with, for example 2 H or 3 H), detection or imaging techniques, such as positron emission tomography (PET) or single- photon emission computed tomography (SPECT) including drug or substrate tissue distribution assays, or in radioactive treatment of patients.
- PET positron emission tomography
- SPECT single- photon emission computed tomography
- an 18 F or labeled compound may be particularly desirable for PET or SPECT studies.
- Isotopically-labeled compounds of formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagents in place of the non-labeled reagent previously employed.
- isotopic enrichment factor means the ratio between the isotopic abundance and the natural abundance of a specified isotope.
- a substituent in a compound of this invention is denoted deuterium, such compound has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium incorporation at each designated deuterium atom), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium incorporation), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5% deuterium incorporation).
- solvates in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D 2 0, de- acetone, d 6 -DMSO.
- Compounds of the invention i.e. compounds of formula (I) that contain groups capable of acting as donors and/or acceptors for hydrogen bonds may be capable of forming co- crystals with suitable co-crystal formers.
- These co-crystals may be prepared from compounds of formula (I) by known co-crystal forming procedures. Such procedures include grinding, heating, co-subliming, co-melting, or contacting in solution compounds of formula (I) with the co-crystal former under crystallization conditions and isolating co- crystals thereby formed.
- Suitable co-crystal formers include those described in WO 2004/078163.
- the invention further provides co-crystals comprising a compound of formula (I).
- the term "pharmaceutically acceptable carrier” includes any and all solvents, dispersion media, coatings, surfactants, antioxidants, preservatives (e.g., antibacterial agents, antifungal agents), isotonic agents, absorption delaying agents, salts, preservatives, drug stabilizers, binders, excipients, disintegration agents, lubricants, sweetening agents, flavoring agents, dyes, and the like and combinations thereof, as would be known to those skilled in the art (see, for example, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289- 1329).
- a therapeutically effective amount of a compound of the present invention refers to an amount of the compound of the present invention that will elicit the biological or medical response of a subject, for example, reduction or inhibition of an enzyme or a protein activity, or ameliorate symptoms, alleviate conditions, slow or delay disease progression, or prevent a disease, etc.
- a therapeutically effective amount refers to the amount of the compound of the present invention that, when administered to a subject, is effective to (1 ) at least partially alleviating, inhibiting, preventing and/or ameliorating a condition, or a disorder or a disease (i) mediated by Btk, or (ii) associated with Btk activity, or (iii) characterized by activity (normal or abnormal) of Btk; or (2) reducing or inhibiting the activity of Btk; or (3) reducing or inhibiting the expression of Btk.
- a therapeutically effective amount refers to the amount of the compound of the present invention that, when administered to a cell, or a tissue, or a non-cellular biological material, or a medium, is effective to at least partially reducing or inhibiting the activity of Btk; or reducing or inhibiting the expression of Btk partially or completely.
- the term "subject" refers to an animal. Typically the animal is a mammal. A subject also refers to for example, primates (e.g., humans, male or female), cows, sheep, goats, horses, dogs, cats, rabbits, rats, mice, fish, birds and the like. In certain embodiments, the subject is a primate. In yet other embodiments, the subject is a human.
- primates e.g., humans, male or female
- the subject is a primate.
- the subject is a human.
- the term “inhibit”, “inhibition” or “inhibiting” refers to the reduction or suppression of a given condition, symptom, or disorder, or disease, or a significant decrease in the baseline activity of a biological activity or process.
- the term “treat”, “treating” or “treatment” of any disease or disorder refers in one embodiment, to ameliorating the disease or disorder (i.e., slowing or arresting or reducing the development of the disease or at least one of the clinical symptoms thereof).
- “treat”, “treating” or “treatment” refers to alleviating or ameliorating at least one physical parameter including those which may not be discernible by the patient.
- “treat”, “treating” or “treatment” refers in one embodiment, to ameliorating the disease or disorder (i.e., slowing or arresting or reducing the development of the disease or at least one of the clinical symptoms thereof).
- “treat”, “treating” or “treatment” refers to alleviating or ameliorating at least one physical parameter including those which may not be discernible by the patient.
- treatment refers to modulating the disease or disorder, either physically, (e.g., stabilization of a discernible symptom), physiologically, (e.g., stabilization of a physical parameter), or both.
- “treat”, “treating” or “treatment” refers to preventing or delaying the onset or development or progression of the disease or disorder.
- a subject is "in need of” a treatment if such subject would benefit biologically, medically or in quality of life from such treatment.
- any asymmetric atom (e.g. , carbon or the like) of the compound(s) of the present invention can be present in racemic or enantiomerically enriched, for example the (R)-, (S)- or (R,S)- configuration.
- each asymmetric atom has at least 50 % enantiomeric excess, at least 60 % enantiomeric excess, at least 70 %
- a compound of the present invention can be in the form of one of the possible isomers, rotamers, atropisomers, tautomers or mixtures thereof, for example, as substantially pure geometric (c/ ' s or trans) isomers, diastereomers, optical isomers (antipodes), racemates or mixtures thereof.
- Any resulting mixtures of isomers can be separated on the basis of the physicochemical differences of the constituents, into the pure or substantially pure geometric or optical isomers, diastereomers, racemates, for example, by chromatography and/or fractional crystallization.
- Any resulting racemates of final products or intermediates can be resolved into the optical antipodes by known methods, e.g., by separation of the diastereomeric salts thereof, obtained with an optically active acid or base, and liberating the optically active acidic or basic compound.
- a basic moiety may thus be employed to resolve the compounds of the present invention into their optical antipodes, e.g., by fractional crystallization of a salt formed with an optically active acid, e.g., tartaric acid, dibenzoyl tartaric acid, diacetyl tartaric acid, di-0,0'-p-toluoyl tartaric acid, mandelic acid, malic acid or camphor-10-sulfonic acid. Racemic products can also be resolved by chiral chromatography, e.g., high pressure liquid chromatography (HPLC) using a chiral adsorbent.
- HPLC high pressure liquid chromatography
- the compounds of the present invention can also be obtained in the form of their hydrates, or include other solvents used for their crystallization.
- the compounds of the present invention may inherently or by design form solvates with pharmaceutically acceptable solvents (including water); therefore, it is intended that the invention embrace both solvated and unsolvated forms.
- solvate refers to a molecular complex of a compound of the present invention
- solvent molecules are those commonly used in the pharmaceutical art, which are known to be innocuous to the recipient, e.g., water, ethanol, and the like.
- solvent molecules are those commonly used in the pharmaceutical art, which are known to be innocuous to the recipient, e.g., water, ethanol, and the like.
- hydrate refers to the complex where the solvent molecule is water.
- the compounds of the present invention including salts, hydrates and solvates thereof, may inherently or by design form polymorphs.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the present invention and a pharmaceutically acceptable carrier.
- the pharmaceutical composition can be formulated for particular routes of administration such as oral administration, parenteral administration, and rectal administration, etc.
- the pharmaceutical compositions of the present invention can be made up in a solid form (including without limitation capsules, tablets, pills, granules, powders or suppositories), or in a liquid form (including without limitation solutions, suspensions or emulsions).
- compositions can be subjected to conventional pharmaceutical operations such as sterilization and/or can contain conventional inert diluents, lubricating agents, or buffering agents, as well as adjuvants, such as preservatives, stabilizers, wetting agents, emulsifiers and buffers, etc.
- the pharmaceutical compositions are tablets or gelatin capsules comprising the active ingredient together with a) diluents, e.g., lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycine; b) lubricants, e.g., silica, talcum, stearic acid, its magnesium or calcium salt and/or polyethyleneglycol; for tablets also c) binders, e.g., magnesium aluminum silicate, starch paste, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone; if desired d) disintegrants, e.g., starches, agar, alginic acid or its sodium salt, or effervescent mixtures; and/or e) absorbents, colorants, flavors and sweeteners.
- diluents e.g., lactose, dextrose, sucrose
- Tablets may be either film coated or enteric coated according to methods known in the art.
- compositions for oral administration include an effective amount of a compound of the invention in the form of tablets, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft capsules, or syrups or elixirs.
- Compositions intended for oral use are prepared according to any method known in the art for the manufacture of pharmaceutical compositions and such compositions can contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets may contain the active ingredient in admixture with nontoxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients are, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example, starch, gelatin or acacia; and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets are uncoated or coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monostearate or glyceryl distearate can be employed.
- Formulations for oral use can be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example, peanut oil, liquid paraffin or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water or an oil medium for example, peanut oil, liquid paraffin or olive oil.
- compositions are aqueous isotonic solutions or suspensions, and suppositories are advantageously prepared from fatty emulsions or suspensions.
- Said compositions may be sterilized and/or contain adjuvants, such as preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers. In addition, they may also contain other therapeutically valuable substances.
- Said compositions are prepared according to conventional mixing, granulating or coating methods, respectively, and contain about 0.1-75%, or contain about 1-50%, of the active ingredient.
- compositions for transdermal application include an effective amount of a compound of the invention with a suitable carrier.
- Carriers suitable for transdermal delivery include absorbable pharmacologically acceptable solvents to assist passage through the skin of the host.
- transdermal devices are in the form of a bandage comprising a backing member, a reservoir containing the compound optionally with carriers, optionally a rate controlling barrier to deliver the compound of the skin of the host at a controlled and predetermined rate over a prolonged period of time, and means to secure the device to the skin.
- compositions for topical application include aqueous solutions, suspensions, ointments, creams, gels or sprayable formulations, e.g., for delivery by aerosol or the like.
- topical delivery systems will in particular be appropriate for dermal application, e.g., for the treatment of skin cancer, e.g., for prophylactic use in sun creams, lotions, sprays and the like. They are thus particularly suited for use in topical, including cosmetic, formulations well-known in the art.
- Such may contain solubilizers, stabilizers, tonicity enhancing agents, buffers and
- a topical application may also pertain to an inhalation or to an intranasal application. They may be conveniently delivered in the form of a dry powder (either alone, as a mixture, for example a dry blend with lactose, or a mixed component particle, for example with phospholipids) from a dry powder inhaler or an aerosol spray presentation from a pressurised container, pump, spray, atomizer or nebuliser, with or without the use of a suitable propellant.
- a dry powder either alone, as a mixture, for example a dry blend with lactose, or a mixed component particle, for example with phospholipids
- the present invention further provides anhydrous pharmaceutical compositions and dosage forms comprising the compounds of the present invention as active ingredients, since water may facilitate the degradation of certain compounds.
- Anhydrous pharmaceutical compositions and dosage forms of the invention can be prepared using anhydrous or low moisture containing ingredients and low moisture or low humidity conditions.
- An anhydrous pharmaceutical composition may be prepared and stored such that its anhydrous nature is maintained. Accordingly, anhydrous compositions are packaged using materials known to prevent exposure to water such that they can be included in suitable formulary kits. Examples of suitable packaging include, but are not limited to, hermetically sealed foils, plastics, unit dose containers (e. g., vials), blister packs, and strip packs.
- compositions and dosage forms that comprise one or more agents that reduce the rate by which the compound of the present invention as an active ingredient will decompose.
- agents which are referred to herein as “stabilizers,” include, but are not limited to, antioxidants such as ascorbic acid, pH buffers, or salt buffers, etc.
- the compounds of formula I in free form or in salt form exhibit valuable pharmacological properties, e.g. Btk modulating properties, e.g. as indicated by in vitro and in vivo tests as provided in the next sections and are therefore indicated for therapy.
- Btk modulating properties e.g. as indicated by in vitro and in vivo tests as provided in the next sections and are therefore indicated for therapy.
- Compounds of the invention may be useful in the treatment of an indication selected from: Autoimmune disorders, inflammatory diseases, allergic diseases, airway diseases, such as asthma and chronic obstructive pulmonary disease (COPD), transplant rejection; diseases in which antibody production, antigen presentation, cytokine production or lymphoid organogenesis are abnormal or are undesirable; including rheumatoid arthritis, systemic onset juvenile idiopathic arthritis (SOJIA), gout, pemphigus vulgaris, idiopathic thrombocytopenic purpura, systemic lupus
- erythematosus erythematosus
- multiple sclerosis myasthenia gravis
- Sjogren's syndrome autoimmune hemolytic anemia
- vasculitides cryoglobulinemia, thrombotic thrombocytopenic purpura, chronic autoimmune urticaria, allergy (atopic dermatitis, contact dermatitis, allergic rhinitis), atherosclerosis, type 1 diabetes, type 2 diabetes, inflammatory bowel disease, ulcerative colitis, morbus Crohn, pancreatitis, glomerolunephritis, Goodpasture's syndrome, Hashimoto's thyroiditis, Grave's disease, antibody-mediated transplant rejection (AMR), graft versus host disease, B cell-mediated hyperacute, acute and chronic transplant rejection; thromboembolic disorders, myocardial infarct, angina pectoris, stroke, ischemic disorders, pulmonary embolism; cancers of haematopoietic origin including but not limited to multiple myeloma; leukemia; acute myelogenous leukemia; chronic myelogenous leukemia; lymphocytic leukemia; myeloid leukemia
- the present invention provides the use of a compound of formula (I) or a salt thereof in therapy.
- the therapy is selected from a disease which may be treated by inhibition of Btk.
- the disease is selected from the afore-mentioned list, suitably rheumatoid arthritis, systemic onset juvenile idiopathic arthritis (SOJIA), pemphigus vulgaris, idiopathic
- thrombocytopenic purpura systemic lupus erythematosus, multiple sclerosis, myasthenia gravis, Sjogren's syndrome, autoimmune hemolytic anemia, anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitides, cryoglobulinemia, thrombotic thrombocytopenic purpura, chronic autoimmune urticaria, atopic dermatitis, allergic rhinitis, type 1 diabetes, type 2 diabetes, inflammatory bowel disease, morbus Crohn, Goodpasture's syndrome, Grave's disease, antibody-mediated transplant rejection (AMR), B cell-mediated hyperacute, acute and chronic transplant rejection; multiple myeloma; acute myelogenous leukemia; chronic myelogenous leukemia; lymphocytic leukemia; myeloid leukemia; non-Hodgkin lymphoma; myelofibrosis with myeloid metaplasia; and Waldenstroem disease, more suitably
- the invention provides a method of treating a disease which is treated by inhibition of Btk kinase comprising administration of a therapeutically acceptable amount of a compound of formula (I) or a salt thereof.
- the disease is selected from the afore-mentioned list, suitably rheumatoid arthritis, systemic onset juvenile idiopathic arthritis (SOJIA), pemphigus vulgaris, idiopathic thrombocytopenic purpura, systemic lupus erythematosus, multiple sclerosis, myasthenia gravis, Sjogren's syndrome, autoimmune hemolytic anemia, anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitides, cryoglobulinemia, thrombotic thrombocytopenic purpura, chronic autoimmune urticaria, atopic dermatitis, allergic rhinitis, type 1 diabetes, type 2 diabetes, inflammatory bowel disease, morbus Crohn,
- SOJIA system
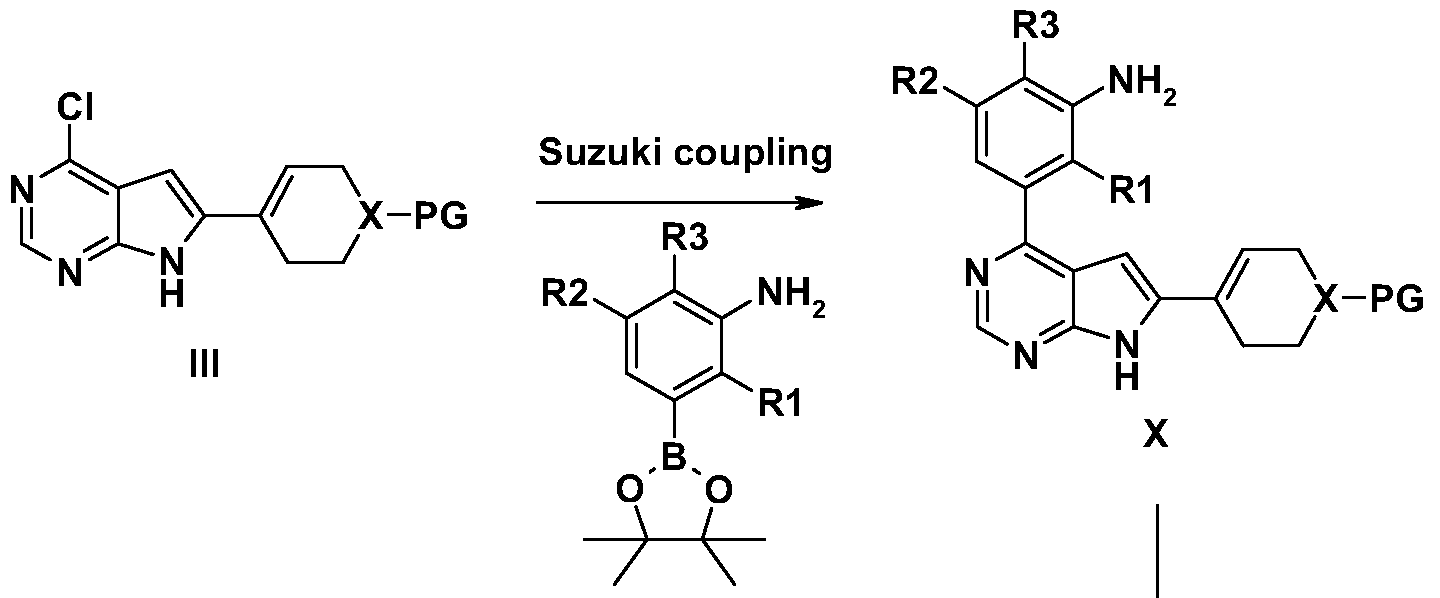
- Agents of the invention may be prepared by a reaction sequence (shown below) involving Suzuki coupling of a protected boronic esters II with the corresponding aryl halides ⁇ , conveniently furnishing intermediate III.
- a reaction sequence shown below
- III e.g. with diluted hydrochloric acid in methanol or the like
- acylation e.g. with an appropriate acetylating agent, e.g.
- the compounds of formula (I) may also be prepared by a reaction sequence involving Suzuki coupling of boronic esters IX with the corresponding aryl halides ⁇ , followed by an additional Suzuki coupling of V with boronic esters VI and an acylation of VII (optionally followed by a deprotection step), as shown in Reaction Scheme 2 below:
- the compounds of formula (I) may also be prepared by a reaction sequence involving Suzuki coupling of boronic esters VI with the corresponding aryl halides III, acylation of intermediate X, followed by deprotection of XI and acylation of XII (optionally followed by a deprotection step), as shown in Reaction Scheme 3 below, wherein X denotes N, and the group PG refers to a protecting group that may be easily removed, such as for example, tert-butyloxycarbonyl.
- the compounds of formula (I) may also be prepared by a reaction sequence involving Suzuki coupling of boronic esters XIII with the corresponding aryl halides III, followed by deprotection of XI and acylation of XII (optionally followed by a deprotection step), as shown in Reaction Scheme 4 below, wherein X denotes N, and the group PG refers to a protecting group that may be easily removed, such as for example, tert-butyloxycarbonyl.
- Reaction Scheme 4 Reaction Scheme 4:
- the compounds of formula (I) may also be prepared by a Suzuki coupling of boronic esters XIV with the corresponding aryl halides III (optionally followed by a deprotection step), as shown in Reaction Scheme 5 below:
- the compounds of formula (I) may also be prepared by urea formation reaction of anilines VII (optionally followed by a deprotection step), as shown in Reaction Scheme 6 below:
- the compounds of formula (I) may be prepared by a reaction sequence involving Suzuki coupling of boronic esters XIII with the corresponding aryl halides XVI. Protection of XVII is followed by halogenation XVII, deprotection of XIX, and Suzuki coupling of halide XX with boronic ester IX (optionally followed by a deprotection step), as shown in Reaction Scheme 7 below, wherein the group PG refers to a protecting group that may be easily removed, such as for example, benzenesulfonyl.
- COMU (1-Cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino- carbenium hexafluorophosphate
- DIPEA Ethyl-diisopropyl-amine, Hunig's base, DIEA
- HATU 0-(7-Azabenzotriazol- 1 -yl)-N , N , N ' , N '- tetramethyluroniumhexafluorophosphat
- TFA Trifluoro-acetic acid
- THF Tetrahydrofuran
- Solvent A Water containing 1 mM ammonium hydrogen carbonate
- Solvent B Acetonitrile containing 0.04% formic acid.
- Solvent A Water containing 0.05% ammonium acetate and 0.05% formic acid.
- Solvent B Acetonitrile containing 0.04% formic acid.
- Example 16 was prepared analogue to Example 37 step 8 by replacing Intermediate 31 with Intermediate 9.
- Example 17 was prepared analogue to Example 16 by replacing Intermediate 8 in step 3 with 4-cyclopropylbenzoic acid.
- Example 18 was prepared analogue to Intermediate 9 by replacing Intermediate 7 with Intermediate 6 and Intermediate 8 with 4-(2,2,2-trifluoro-1-hydroxy-1-methyl-ethyl)- benzoic acid (WO2007/145834).
- Example 19 was prepared analogue to Example 18 by replacing 4-(2,2,2-trifluoro-1- hydroxy-1-methyl-ethyl)-benzoic acid with 4-acetyl-benzoic acid.
- Example 21 was prepared analogue to Intermediate 9 by replacing Intermediate 7 with Intermediate 10 and Intermediate 8 with 4-(2-hydroxy-1 ,1-dimethyl-ethyl)-benzoic acid.
- Intermediate 11 was prepared analogue to Intermediate 1 by replacing 4-(4, 4,5,5- tetramethyl-[1 ,2,3]dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylic acid tert-butyl ester with 3,6-dihydro-2H-pyran-4-boronic acid pinacolester.
- Example 22 was prepared analogue to Intermediate 9 by replacing Intermediate 7 with Intermediate 12.
- Example 23 was prepared analogue to Example 16 step 4 by replacing Intermediate 9 with Intermediate 14.
- Example 24 was prepared analogue to Example 15 by replacing Intermediate 6 with Intermediate 16 and 4-dimethyaminobenzoyl chloride with tert-butylbenzoyl chloride.
- Example 24 (160 mg, 0.32 mmol) was dissolved in acetic acid (5 ml) and hydrogen peroxide (0.033 ml, 0.32 mmol) was added. The mixture was stirred at room temperature for 2 hours. The mixture was treated with sodium hydrogen sulfite solution (10%, 10 ml) for 10 minutes, diluted with water, basified with 2N sodium hydroxide solution and extracted with EtOAc. The organic layer was washed with brine (2x), dried over sodium sulfate, filtered and evaporated.
- Example 24 (160 mg, 0.32 mmol) was dissolved in DCM (15 ml), then trifluoro acetic acid (5 ml) and hydrogen peroxide (0.065 ml, 0.64 mmol) were added. The mixture was stirred at room temperature for 2 hours. The mixture was treated with sodium hydrogen sulfite solution (10%, 10 ml) for 10 minutes, diluted with water, basified with 2N sodium hydroxide solution and extracted with EtOAc. The organic layer was washed with brine (2x), dried over sodium sulfate, filtered and evaporated.
- Intermediate 17 was prepared analogue to Intermediate 1 by replacing 4-(4, 4,5,5- tetramethyl-[1 ,2,3]dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylic acid tert-butyl ester with 1 ,4-dioxaspiro[5,5]dec-7-en-8-boronic acid pinacol ester.
- Example 27 was prepared analogue to Example 15 by replacing Intermediate 6 with Intermediate 18 and 4-dimethyaminobenzoyl chloride with 4-tert-butylbenzoyl chloride.
- Example 27 A mixture of Example 27 (450 mg, 0.832 mmol) and TFA (6 ml) in DCM (30ml) was stirred at r.t. for 6 hrs. The solvents were removed in vacuo and the crude product was purified by flash chromatography (silica gel, EtOAc/MeOH/NH 4 OH gradient) to yield Intermediate 19.
- Example 34 was prepared analogue to Example 30 by replacing Intermediate 22 with Intermediate 24.
- Example 35 was prepared analogue to Example 31 by replacing Intermediate 22 with Intermediate 24.
- Example 36 was prepared analogue to Example 32 by replacing Example 31 with Example 35.
- Example 38 was prepared analogue to Example 37 step 8 by replacing Intermediate 31 with Intermediate 32.
- Example 39 was prepared analogue to Example 37 by replacing the boronic ester Intermediate 28 in step 5 with the boronic ester Intermediate 36.
- Example 40 was prepared analogue to Intermediate 30 in Example 37 step 6 by replacing Intermediate 28 in Example 37 step 5 with Intermediate 38.
- Example 41 was prepared analogue to Example 37 step 7 by replacing Intermediate 30 with Example 40.
- Example 40 To a solution of Example 40 (90 mg, 0.186 mmol) and DIPEA (0.098 ml, 0.558 mmol) in THF (10 ml) was added methanesulfonyl chloride (0.015 ml, 0.186 mmol) dropwise. The resulting mixture was stirred at r.t. for 1 hr, then quenched with water and diluted with EtOAc. The organic layer was washed with sat. aqueous NaHC0 3 solution and brine, dried with Na 2 S0 4 , and filtered. The solvents were removed in vacuo, and the crude product was purified by reversed phase HPLC (MeCN/H 2 0 gradient) to yield Example 42 as a light yellow solid.
- Methanesulfonyl chloride 0.015 ml, 0.186 mmol
- Example 43 was prepared analogue to Example 42 by replacing methanesulfonyl chloride with ⁇ , ⁇ -dimethylamidosulfamoyl chloride.
- Example 45 was prepared analogue to Intermediate 6 by replacing Intermediate 5 with Intermediate 40.
- Example 46 was prepared analogue to Intermediate 6 by replacing Intermediate 5 with Intermediate 41.
- Example 47 was prepared analogue to Intermediate 6 by replacing Intermediate 5 with Intermediate 45, followed by basic removal of the acetate protecting group with LiOH in MeOH/water.
- Example 48 was prepared analogue to Intermediate 29 by replacing Intermediate 1 with Intermediate 11 , followed by removal of the TBDPS protecting group with TBAF in THF.
- Example 49 was prepared analogue to Intermediate 6 by replacing Intermediate 3 with Intermediate 11 and Intermediate 5 with Intermediate 38.
- Example 50 was prepared analogue to Example 49 by replacing Intermediate 38 with Intermediate 47.
- Example 51 For the Suzuki coupling between chloride Intermediate 15 (0.25 g, 0.99 mmol) and the boronic ester Intermediate 48 (0.78 g, 1.98 mmol) the same protocol was used as described in Example 1 step 1 to afford Example 51 as a beige solide.
- Example 51 Compound of Example 51 (250 mg, 0.51 mmol) was dissolved in DCM (5 ml), then trifluoro acetic acid (5 ml) and hydrogen peroxide (0.079 ml, 0.78 mmol) were added. The mixture was stirred at room temperature for 2 hours. The mixture was treated with sodium hydrogen sulfite solution 10 % (10 ml) for 10 minutes, diluted with water, basified with 2N sodium hydroxide solution and extracted with EtOAc. The organic layer was washed with brine (2x), dried over sodium sulfate, filtered and evaporated. The residue was purified by flash chromatography on silica (EtOAc to EtOAc/MeOH/NH 4 OH 98:2:0.2) to afford the compound Example 53 as a beige solid.
- Example 54 was prepared analogue to Intermediate 29 by replacing Intermediate 1 with Intermediate 17, followed by removal of the TBDPS protecting group with TBAF in THF.
- Example 57 was purified by flash chromatography (siliga gel, EtOAc/MeOH gradient) to afford Example 57 as beige solid.
- Example 58 was prepared analogue to Example 57 by replacing 3-tert-butoxy-azetidine hydrochloride with 3-isopropoxy-azetidine hydrochloride.
- Example 59 was prepared analogue to Example 57 by replacing 3-tert-butoxy-azetidine hydrochloride with 5-fluoro-2,3-dihydro-1 H-isoindole.
- Example 60 was prepared analogue to Example 57 by replacing 3-tert-butoxy-azetidine hydrochloride with 3-(2,2,2-trifluoro-1-trifluoromethyl-ethoxy)-azetidine hydrochloride (WO2009/077334).
- Example 61 was prepared analogue to Example 57 by replacing Intermediate 6 with Intermediate 12.
- Example 62 was prepared analogue to Example 57 by replacing Intermediate 6 with Intermediate 10.
- Intermediate 10 was prepared analogue to Intermediate 6 by replacing Intermediate 5 with 5-fluoro-2-methyl-3-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)aniline.
- Example 62 was prepared analogue to Example 57 by replacing Intermediate 6 with Intermediate 50.
- Example 64 was prepared analogue to Intermediate 11 by replacing 4-chloro-6-iodo- 7H-pyrrolo[2,3-d]pyrimidine with Intermediate 54.
- Example 65 was prepared analogue to Intermediate 17 by replacing 4-chloro-6-iodo- 7H-pyrrolo[2,3-d]pyrimidine with Intermediate 54.
- Example 66 was prepared analogue to Example 28 step 2 by replacing Intermediate 19 in with Intermediate 55.
- Example 67 was prepared analogue to Example 56 by replacing Intermediate 49 with Intermediate 55.
- the inhibitory activity of the present compounds against Btk was assessed in a biochemical enzyme assay.
- Assay plates in 384 well format were prepared with 8-point serial dilutions for the test compounds on a Thermo CatX workstation equipped with a Innovadyne Nanodrop Express.
- the assay plates were prepared by addition of 50 nl per well of compound solution in 90 % DMSO.
- kinase reactions were started by stepwise addition of 4.5 ⁇ per well of peptide/ATP-solution (4 ⁇ FITC-Ahx- TSELKKVVALYDYMPMNAND-NH2, 164 ⁇ ATP) in kinase buffer (50mM HEPES, pH 7.5, 1 mM DTT, 0.02% Tween20, 0.02% BSA, 0.6% DMSO, 10 mM beta- glycerophosphate, and 10 ⁇ sodium orthovanadate, 18 mM MgCI2, 1 mM MnCI2) and 4.5 ⁇ per well of enzyme solution (6.4nM full-lenght human recombinant BTK) in kinase buffer.
- peptide/ATP-solution 4 ⁇ FITC-Ahx- TSELKKVVALYDYMPMNAND-NH2, 164 ⁇ ATP
- kinase buffer 50mM HEPES, pH 7.5, 1 mM DTT, 0.02% Tween
- Kinase reactions were incubated at 30°C for 60 minutes and subsequently terminated by addition of 16 ⁇ per well of stop solution (100 mM HEPES pH 7.5, 5 % DMSO, 0.1 % Caliper coating reagent, 10 mM EDTA, and 0.015 % Brij35).
- Stop solution 100 mM HEPES pH 7.5, 5 % DMSO, 0.1 % Caliper coating reagent, 10 mM EDTA, and 0.015 % Brij35.
- Kinase reactions were analyzed on a Caliper LC3000 workstation by separating phosphorylated and unphosphorylated peptides and kinase activities were calculated from the amounts of newly formed phospho-peptide.
- Inhibition data were calculated by comparison to control reactions without enzyme (100 % inhibition) and without inhibitors (0 % inhibition). The concentration of inhibitor required for 50 % inhibition (IC50) was calculated from the inhibition in response to inhibitor concentrations.
- Example 44 0.017
- Example 45 0.008
- the present compounds might also be assessed for their capacity to inhibit Btk-dependent FcG receptor-induced IL-8 secretion in human cells.
- the human myeloid leukemia THP1 cell line (ATCC TIB202) was grown in RPMI 1640 medium
- tissue-culture grade 384-well plates was coated with human IgG of unknown specificity by incubating overnight at 4°C with 40 ⁇ /well of a 50 ⁇ g/ml IgG solution in PBS. On the day of the experiment, plates were washed 5 times with 80 ⁇ water on a Molecular Devices Aquamax DW4 plate washer. Solutions of the test compounds in 90 % DMSO were added to each well on a Hamilton Microlab Star liquid handling station to 40 ⁇ /well tissue culture medium and the total DMSO concentration was adjusted to 0.1 %.
- Differentiated THP1 cells were then added in 40 ⁇ /well to reach a final density of 5 ⁇ 00 cells/well in 80 ⁇ culture medium. After 24 hours, IL-8 secretion was measured in the supernatant by the IL-8 HTRF assay following the protocol of the vendor (CisBio international). Inhibition data were calculated by comparison to control cultures without IgG stimulus enzyme (100 % inhibition) and without inhibitors (0 % inhibition). The concentration of inhibitor required for 50 % inhibition (IC50) was calculated from the inhibition in response to inhibitor concentrations.
- the inhibitory activity of the present compounds in blood was assessed in the following in vitro B cell activation assay.
- Whole blood was collected from the abdominal aorta of anaesthetized adult male Lewis rats and was anticoagulated with 100 U/ml sodium heparin. Blood was then diluted to 50 % with high glucose DMEM
- compounds of the invention may generally be useful in the treatment of an indication selected from:
- Autoimmune disorders inflammatory diseases, allergic diseases, airway diseases, such as asthma and chronic obstructive pulmonary disease (COPD), transplant rejection; diseases in which antibody production, antigen presentation, cytokine production or lymphoid organogenesis are abnormal or are undesirable; including rheumatoid arthritis, systemic onset juvenile idiopathic arthritis (SOJIA), gout, pemphigus vulgaris, idiopathic thrombocytopenic purpura, systemic lupus erythematosus, multiple sclerosis, myasthenia gravis, Sjogren's syndrome, autoimmune hemolytic anemia, anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitides, cryoglobulinemia, thrombotic thrombocytopenic purpura, chronic autoimmune urticaria, allergy (atopic dermatitis, contact dermatitis, allergic rhinitis), atherosclerosis, type 1 diabetes, type 2 diabetes, inflammatory bowel disease, ulcer
- glomerolunephritis Goodpasture's syndrome, Hashimoto's thyroiditis, Grave's disease, antibody-mediated transplant rejection (AMR), graft versus host disease, B cell-mediated hyperacute, acute and chronic transplant rejection; thromboembolic disorders, myocardial infarct, angina pectoris, stroke, ischemic disorders, pulmonary embolism; cancers of haematopoietic origin including but not limited to multiple myeloma; a leukaemia; acute myelogenous leukemia; chronic myelogenous leukemia; lymphocytic leukemia; myeloid leukemia; non-Hodgkin lymphoma; lymphomas; polycythemia vera; essential thrombocythemia; myelofibrosis with myeloid metaplasia; and Waldenstroem disease.
- AMR antibody-mediated transplant rejection
- graft versus host disease B cell-mediated hyperacute, acute and chronic transplant rejection
- the therapy is selected from a disease which may be treated by an antagonist of Bruton's tyrosine kinase.
- the invention provides a method of treating a disease which is treated by the modulation of Btk- comprising administration of a therapeutically acceptable amount of a compound of formula (I) or a salt thereof.
- the disease is selected from the afore-mentioned lists
- the compound of the present invention may be administered either simultaneously with, or before or after, one or more other therapeutic agent.
- the compound of the present invention may be administered separately, by the same or different route of
- the compounds of formula (I) may be administered as the sole active ingredient or in conjunction with, e.g. as an adjuvant to, other drugs e.g. immunosuppressive or immunomodulating agents or other anti-inflammatory agents, e.g. for the treatment or prevention of alio- or xenograft acute or chronic rejection or inflammatory or autoimmune disorders, or a chemotherapeutic agent, e.g a malignant cell anti-proliferative agent.
- the compounds of formula (I) may be used in combination with a calcineurin inhibitor, e.g. cyclosporin A or FK 506; a mTOR inhibitor, e.g.
- rapamycin 40-O-(2- hydroxyethyl)-rapamycin, CCI779, ABT578, AP23573, AP23464, AP23675, AP23841 , TAFA-93, biolimus-7 or biolimus-9; an ascomycin having immunosuppressive properties, e.g. ABT-281 , ASM981 , etc.; corticosteroids; cyclophosphamide; azathioprene; methotrexate; leflunomide; mizoribine; mycophenolic acid or salt; mycophenolate mofetil; 15-deoxyspergualine or an immunosuppressive homologue, analogue or derivative thereof; a PKC inhibitor, e.g.
- a JAK3 kinase inhibitor e.g. N-benzyl-3,4-dihydroxy- benzylidene-cyanoacetamide a-cyano-(3,4-dihydroxy)-]N-benzylcinnamamide
- mono-citrate also called CP-690,550
- sphingosine-1-phosphate receptor modulators such as FTY720 (fingolimod), or compounds disclosed in WO 2005/000833
- immunosuppressive monoclonal antibodies e.g., monoclonal antibodies to leukocyte receptors, e.g., MHC, CD2, CD3, CD4, CD7, CD8, CD25, CD28, CD40, CD45, CD52, CD58, CD80, CD86 or their ligands
- other immunomodulatory compounds e.g.
- a recombinant binding molecule having at least a portion of the extracellular domain of CTLA4 or a mutant thereof, e.g. an at least extracellular portion of CTLA4 or a mutant thereof joined to a non-CTLA4 protein sequence, e.g. CTLA4lg (for ex. designated ATCC 68629) or a mutant thereof, e.g. LEA29Y; adhesion molecule inhibitors, e.g. LFA- 1 antagonists, ICAM-1 or -3 antagonists, VCAM-4 antagonists or VLA-4 antagonists; or a chemotherapeutic agent, e.g.
- a compound of formula (I) may be selected from a PI3K inhibitor (e.g. pan, or alpha, beta, gamma, delta selectives), TNF inhibitors, ILI beta inhibitors, IL17 inhibitors, and inhibitors of IL6 or IL receptor.
- co-administration or “combined administration” or the like as utilized herein are meant to encompass administration of the selected therapeutic agents to a single patient, and are intended to include treatment regimens in which the agents are not necessarily administered by the same route of administration or at the same time.
- pharmaceutical combination as used herein means a product that results from the mixing or combining of more than one active ingredient and includes both fixed and non-fixed combinations of the active ingredients.
- fixed combination means that the active ingredients, e.g. a compound of formula (I) and a co-agent, are both administered to a patient simultaneously in the form of a single entity or dosage.
- non-fixed combination means that the active ingredients, e.g.
- a compound of formula (I) and a co-agent are both administered to a patient as separate entities either simultaneously, concurrently or sequentially with no specific time limits, wherein such administration provides therapeutically effective levels of the 2 compounds in the body of the patient.
- cocktail therapy e.g. the administration of 3 or more active ingredients.
- the invention provides a product comprising a compound of formula (I) and at least one other therapeutic agent as a combined preparation for simultaneous, separate or sequential use in therapy.
- the therapy is the treatment of a disease or condition mediated by Btk kinases.
- Products provided as a combined preparation include a composition comprising the compound of formula (I) and the other therapeutic agent(s) together in the same pharmaceutical composition, or the compound of formula (I) and the other therapeutic agent(s) in separate form, e.g. in the form of a kit.
- the invention provides a pharmaceutical composition comprising a compound of formula (I) and another therapeutic agent(s).
- a pharmaceutical composition comprising a compound of formula (I) and another therapeutic agent(s).
- composition may comprise a pharmaceutically acceptable excipient, as described above.
- the invention provides a kit comprising two or more separate pharmaceutical compositions, at least one of which contains a compound of formula (I).
- the kit comprises means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet.
- a container, divided bottle, or divided foil packet An example of such a kit is a blister pack, as typically used for the packaging of tablets, capsules and the like.
- the kit of the invention may be used for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another.
- the kit of the invention typically comprises directions for
- the compound of the invention and the other therapeutic agent may be manufactured and/or formulated by the same or different manufacturers. Moreover, the compound of the invention and the other therapeutic may be brought together into a combination therapy: (i) prior to release of the combination product to physicians (e.g. in the case of a kit comprising the compound of the invention and the other therapeutic agent); (ii) by the physician themselves (or under the guidance of the physician) shortly before administration; (iii) in the patient themselves, e.g. during sequential administration of the compound of the invention and the other therapeutic agent.
- the invention provides the use of a compound of formula (I) for treating a disease or condition mediated by Btk kinases, wherein the medicament is prepared for administration with another therapeutic agent.
- the invention also provides the use of another therapeutic agent for treating a disease or condition mediated by Btk , wherein the medicament is administered with a compound of formula (I).
- the invention also provides a compound of formula (I) for use in a method of treating a disease or condition mediated by Btk, wherein the compound of formula (I) is prepared for administration with another therapeutic agent.
- the invention also provides another therapeutic agent for use in a method of treating a disease or condition mediated by Btk , wherein the other therapeutic agent is prepared for administration with a compound of formula (I).
- the invention also provides a compound of formula (I) for use in a method of treating a disease or condition mediated by Btk , wherein the compound of formula (I) is administered with another therapeutic agent.
- the invention also provides another therapeutic agent for use in a method of treating a disease or condition mediated by Btk, wherein the other therapeutic agent is administered with a compound of formula (I).
Abstract
Description































































































































































Claims




Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES12761655.5T ES2548414T3 (en) | 2011-07-08 | 2012-07-07 | Novel pyrimidine pyrrolo derivatives |
| EA201490229A EA201490229A1 (en) | 2011-07-08 | 2012-07-07 | NEW PYRROPHYRIMIDINE DERIVATIVES |
| JP2014517980A JP6145451B2 (en) | 2011-07-08 | 2012-07-07 | Novel pyrrolopyrimidine derivatives |
| KR1020147002986A KR20140058543A (en) | 2011-07-08 | 2012-07-07 | Novel pyrrolo pyrimidine derivatives |
| EP12761655.5A EP2729466B1 (en) | 2011-07-08 | 2012-07-07 | Novel pyrrolo pyrimidine derivatives |
| AU2012282229A AU2012282229B2 (en) | 2011-07-08 | 2012-07-07 | Novel pyrrolo pyrimidine derivatives |
| CN201280033935.9A CN103732596B (en) | 2011-07-08 | 2012-07-07 | Pyrrolopyrimidine derivatives |
| MX2014000338A MX2014000338A (en) | 2011-07-08 | 2012-07-07 | Novel pyrrolo pyrimidine derivatives. |
| CA2841111A CA2841111A1 (en) | 2011-07-08 | 2012-07-07 | Novel pyrrolo pyrimidine derivatives |
| US14/130,536 US9233111B2 (en) | 2011-07-08 | 2012-07-07 | Pyrrolo pyrimidine derivatives |
| BR112014000314A BR112014000314A2 (en) | 2011-07-08 | 2012-07-07 | pyrrole pyrimidine derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161505560P | 2011-07-08 | 2011-07-08 | |
| US61/505,560 | 2011-07-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013008095A1 true WO2013008095A1 (en) | 2013-01-17 |
Family
ID=46880751
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2012/001699 WO2013008095A1 (en) | 2011-07-08 | 2012-07-07 | Novel pyrrolo pyrimidine derivatives |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US9233111B2 (en) |
| EP (1) | EP2729466B1 (en) |
| JP (1) | JP6145451B2 (en) |
| KR (1) | KR20140058543A (en) |
| CN (1) | CN103732596B (en) |
| AU (1) | AU2012282229B2 (en) |
| BR (1) | BR112014000314A2 (en) |
| CA (1) | CA2841111A1 (en) |
| EA (1) | EA201490229A1 (en) |
| ES (1) | ES2548414T3 (en) |
| MX (1) | MX2014000338A (en) |
| WO (1) | WO2013008095A1 (en) |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013157022A1 (en) | 2012-04-20 | 2013-10-24 | Advinus Therapeutics Limited | Substituted hetero-bicyclic compounds, compositions and medicinal applications thereof |
| WO2013157021A1 (en) | 2012-04-20 | 2013-10-24 | Advinus Therapeutics Limited | Bicyclic compounds, compositions and medicinal applications thereof |
| US9233111B2 (en) | 2011-07-08 | 2016-01-12 | Novartis Ag | Pyrrolo pyrimidine derivatives |
| EP3042903A1 (en) | 2015-01-06 | 2016-07-13 | Advinus Therapeutics Limited | Substituted hetero-bicyclic compounds, compositions and medicinal applications thereof |
| US9512084B2 (en) | 2013-11-29 | 2016-12-06 | Novartis Ag | Amino pyrimidine derivatives |
| WO2017106634A1 (en) * | 2015-12-17 | 2017-06-22 | Incyte Corporation | N-phenyl-pyridine-2-carboxamide derivatives and their use as pd-1/pd-l1 protein/protein interaction modulators |
| WO2018053437A1 (en) | 2016-09-19 | 2018-03-22 | Mei Pharma, Inc. | Combination therapy |
| US10308644B2 (en) | 2016-12-22 | 2019-06-04 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2019186358A1 (en) | 2018-03-26 | 2019-10-03 | Novartis Ag | 3-hydroxy-n-(3-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)pyrrolidine-1-carboxamide derivatives |
| WO2019186343A1 (en) | 2018-03-26 | 2019-10-03 | Novartis Ag | N-(3-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)benzamide derivatives |
| EP3550031A1 (en) | 2012-07-24 | 2019-10-09 | Pharmacyclics, LLC | Mutations associated with resistance to inhibitors of bruton's tyrosine kinase (btk) |
| EP3556761A4 (en) * | 2017-02-08 | 2019-12-25 | The National Institutes Of Pharmaceutical Research | Pyrrolo-aromatic heterocyclic compound, preparation method therefor, and medical use thereof |
| US10618916B2 (en) | 2018-05-11 | 2020-04-14 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10669271B2 (en) | 2018-03-30 | 2020-06-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| JP2020105181A (en) * | 2013-12-02 | 2020-07-09 | ファーマサイクリックス エルエルシー | Methods of treating and preventing alloantibody driven chronic graft versus host disease |
| US10793565B2 (en) | 2016-12-22 | 2020-10-06 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10806785B2 (en) | 2016-12-22 | 2020-10-20 | Incyte Corporation | Immunomodulator compounds and methods of use |
| US11401279B2 (en) | 2019-09-30 | 2022-08-02 | Incyte Corporation | Pyrido[3,2-d]pyrimidine compounds as immunomodulators |
| US11407749B2 (en) | 2015-10-19 | 2022-08-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2022189859A1 (en) | 2021-03-12 | 2022-09-15 | Novartis Ag | Fatty acid-bifunctional degrader conjugates and their methods of use |
| US11465981B2 (en) | 2016-12-22 | 2022-10-11 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11535615B2 (en) | 2015-12-22 | 2022-12-27 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11572366B2 (en) | 2015-11-19 | 2023-02-07 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11608337B2 (en) | 2016-05-06 | 2023-03-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11613536B2 (en) | 2016-08-29 | 2023-03-28 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11623921B2 (en) | 2014-10-24 | 2023-04-11 | Bristol-Myers Squibb Company | Indole carboxamide compounds |
| US11673883B2 (en) | 2016-05-26 | 2023-06-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11718605B2 (en) | 2016-07-14 | 2023-08-08 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11753406B2 (en) | 2019-08-09 | 2023-09-12 | Incyte Corporation | Salts of a PD-1/PD-L1 inhibitor |
| US11760756B2 (en) | 2020-11-06 | 2023-09-19 | Incyte Corporation | Crystalline form of a PD-1/PD-L1 inhibitor |
| US11780836B2 (en) | 2020-11-06 | 2023-10-10 | Incyte Corporation | Process of preparing a PD-1/PD-L1 inhibitor |
| US11866434B2 (en) | 2020-11-06 | 2024-01-09 | Incyte Corporation | Process for making a PD-1/PD-L1 inhibitor and salts and crystalline forms thereof |
| US11866451B2 (en) | 2019-11-11 | 2024-01-09 | Incyte Corporation | Salts and crystalline forms of a PD-1/PD-L1 inhibitor |
| US11873309B2 (en) | 2016-06-20 | 2024-01-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ711540A (en) | 2013-04-25 | 2018-08-31 | Beigene Ltd | Fused heterocyclic compounds as protein kinase inhibitors |
| EP4130044A1 (en) | 2013-09-13 | 2023-02-08 | BeiGene Switzerland GmbH | Anti-pd1 antibodies and their use as therapeutics and diagnostics |
| TWI726608B (en) | 2014-07-03 | 2021-05-01 | 英屬開曼群島商百濟神州有限公司 | Anti-pd-l1 antibodies and their use as therapeutics and diagnostics |
| NZ729603A (en) * | 2014-10-06 | 2022-02-25 | Merck Patent Gmbh | Heteroaryl compounds as btk inhibitors and uses thereof |
| WO2016079669A1 (en) * | 2014-11-19 | 2016-05-26 | Novartis Ag | Labeled amino pyrimidine derivatives |
| AR102871A1 (en) * | 2014-12-03 | 2017-03-29 | Pharmacyclics Llc | FIBROSIS TREATMENT METHODS |
| AU2017293423B2 (en) | 2016-07-05 | 2023-05-25 | Beigene, Ltd. | Combination of a PD-1 antagonist and a RAF inhibitor for treating cancer |
| CN109563099B (en) | 2016-08-16 | 2023-02-03 | 百济神州有限公司 | Crystal form of compound, preparation and application thereof |
| WO2018033135A1 (en) | 2016-08-19 | 2018-02-22 | Beigene, Ltd. | Use of a combination comprising a btk inhibitor for treating cancers |
| US11555038B2 (en) | 2017-01-25 | 2023-01-17 | Beigene, Ltd. | Crystalline forms of (S)-7-(1-(but-2-ynoyl)piperidin-4-yl)-2-(4-phenoxyphenyl)-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine-3-carboxamide, preparation, and uses thereof |
| AU2018290532A1 (en) | 2017-06-26 | 2019-11-21 | Beigene, Ltd. | Immunotherapy for hepatocellular carcinoma |
| CN110997677A (en) | 2017-08-12 | 2020-04-10 | 百济神州有限公司 | Btk inhibitors with improved dual selectivity |
| WO2019108795A1 (en) | 2017-11-29 | 2019-06-06 | Beigene Switzerland Gmbh | Treatment of indolent or aggressive b-cell lymphomas using a combination comprising btk inhibitors |
| CN111704617B (en) * | 2020-06-15 | 2022-08-23 | 嘉兴特科罗生物科技有限公司 | Small molecule compound |
| CN115073469B (en) * | 2021-03-15 | 2023-12-22 | 药雅科技(上海)有限公司 | Preparation and application of pyrrolopyrimidine compound as kinase inhibitor |
| US11786531B1 (en) | 2022-06-08 | 2023-10-17 | Beigene Switzerland Gmbh | Methods of treating B-cell proliferative disorder |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002038561A1 (en) | 2000-11-07 | 2002-05-16 | Novartis Ag | Indolylmaleimide derivatives as protein kinase c inhibitors |
| WO2003082859A1 (en) | 2002-04-03 | 2003-10-09 | Novartis Ag | Indolylmaleimide derivatives |
| WO2004052359A1 (en) | 2002-12-09 | 2004-06-24 | The Board Of Regents Of The University Of Texas System | Methods for selectively inhibiting janus tyrosine kinase 3 (jak3) |
| WO2004078163A2 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2005000833A1 (en) | 2003-05-19 | 2005-01-06 | Irm, Llc | Immunosuppressant compounds and compositions |
| WO2005066156A1 (en) | 2004-01-12 | 2005-07-21 | Cytopia Research Pty Ltd | Selective kinase inhibitors |
| WO2007090752A1 (en) | 2006-02-07 | 2007-08-16 | F. Hoffmann-La Roche Ag | Anthranilamide/2-amino-heteroarene carboxamide derivatives |
| WO2007145834A2 (en) | 2006-06-08 | 2007-12-21 | Amgen Inc. | Benzamide derivatives and uses related thereto |
| US20090105209A1 (en) | 2007-10-23 | 2009-04-23 | Roche Palo Alto Llc | BTK protein kinase inhibitors |
| WO2009077334A1 (en) | 2007-12-14 | 2009-06-25 | F. Hoffmann-La Roche Ag | Novel imidazo[1,2-a]pyridine and imidazo[1,2-b]pyridazine derivatives |
| US20090306041A1 (en) | 2008-02-05 | 2009-12-10 | Roche Palo Alto Llc | Inhibitors of Bruton's tyrosine kinase |
| WO2010000633A1 (en) | 2008-07-02 | 2010-01-07 | F. Hoffmann-La Roche Ag | Novel phenylpyrazinones as kinase inhibitors |
Family Cites Families (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6303652B1 (en) | 1998-08-21 | 2001-10-16 | Hughes Institute | BTK inhibitors and methods for their identification and use |
| SK18542000A3 (en) | 1998-06-04 | 2001-12-03 | Abbott Laboratories | Cell adhesion-inhibiting anti-inflammatory compounds |
| PA8474101A1 (en) | 1998-06-19 | 2000-09-29 | Pfizer Prod Inc | PYROLEUM [2,3-D] PIRIMIDINE COMPOUNDS |
| GB2345868A (en) | 1999-01-23 | 2000-07-26 | Martin Hodgson | Self adhesive magnetic or electromagnetic tapes |
| WO2000075145A1 (en) | 1999-06-03 | 2000-12-14 | Abbott Laboratories | Cell adhesion-inhibiting antiinflammatory compounds |
| GB0005345D0 (en) | 2000-03-06 | 2000-04-26 | Mathilda & Terence Kennedy Ins | Methods of treating sepsis septic shock and inflammation |
| WO2002038797A2 (en) | 2000-10-23 | 2002-05-16 | Bristol-Myers Squibb Company | Modulators of bruton's tyrosine kinase, their identification and use |
| EP1217000A1 (en) | 2000-12-23 | 2002-06-26 | Aventis Pharma Deutschland GmbH | Inhibitors of factor Xa and factor VIIa |
| GB0119249D0 (en) * | 2001-08-07 | 2001-10-03 | Novartis Ag | Organic compounds |
| EP2322201A3 (en) | 2002-10-29 | 2011-07-27 | Genentech, Inc. | Compositions and methods for the treatment of immune related diseases |
| EP1473039A1 (en) | 2003-05-02 | 2004-11-03 | Centre National De La Recherche Scientifique (Cnrs) | Use of inhibitors and antisense oligonucleotides of BTK for the treatment of proliferative mastocytosis |
| US7393848B2 (en) | 2003-06-30 | 2008-07-01 | Cgi Pharmaceuticals, Inc. | Certain heterocyclic substituted imidazo[1,2-A]pyrazin-8-ylamines and methods of inhibition of Bruton's tyrosine kinase by such compounds |
| PE20051092A1 (en) * | 2004-01-29 | 2006-01-20 | Novartis Ag | PYROLO-PYRIMIDINE DERIVATIVES USEFUL FOR THE TREATMENT OF PROLIFERATIVE DISEASES |
| JP2008508358A (en) | 2004-08-02 | 2008-03-21 | オーエスアイ・ファーマスーティカルズ・インコーポレーテッド | Aryl-amino substituted pyrrolopyrimidine multikinase inhibitor compounds |
| GB0420719D0 (en) | 2004-09-17 | 2004-10-20 | Addex Pharmaceuticals Sa | Novel allosteric modulators |
| WO2006099075A2 (en) | 2005-03-10 | 2006-09-21 | Cgi Pharmaceuticals, Inc. | Certain substituted amides, method of making, and method of use thereof |
| WO2006101783A2 (en) | 2005-03-15 | 2006-09-28 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| MX2007014066A (en) | 2005-05-13 | 2008-02-11 | Irm Llc | Compounds and compositions as protein kinase inhibitors. |
| CA2608726C (en) | 2005-05-20 | 2013-07-09 | Methylgene Inc. | Inhibitors of vegf receptor and hgf receptor signaling |
| EP1963320A1 (en) * | 2005-12-07 | 2008-09-03 | OSI Pharmaceuticals, Inc. | Pyrrolopyridine kinase inhibiting compounds |
| TW201434835A (en) | 2005-12-13 | 2014-09-16 | Incyte Corp | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| US7915411B2 (en) | 2005-12-21 | 2011-03-29 | Abbott Laboratories | Anti-viral compounds |
| US20070208053A1 (en) | 2006-01-19 | 2007-09-06 | Arnold Lee D | Fused heterobicyclic kinase inhibitors |
| TWI398252B (en) | 2006-05-26 | 2013-06-11 | Novartis Ag | Pyrrolopyrimidine compounds and their uses |
| DE102006033109A1 (en) | 2006-07-18 | 2008-01-31 | Grünenthal GmbH | Substituted heteroaryl derivatives |
| US20100160292A1 (en) | 2006-09-11 | 2010-06-24 | Cgi Pharmaceuticals, Inc | Kinase Inhibitors, and Methods of Using and Identifying Kinase Inhibitors |
| AR063946A1 (en) | 2006-09-11 | 2009-03-04 | Cgi Pharmaceuticals Inc | CERTAIN REPLACED PIRIMIDINS, THE USE OF THE SAME FOR THE TREATMENT OF DISEASES MEDIATED BY THE INHIBITION OF THE ACTIVITY OF BTK AND PHARMACEUTICAL COMPOSITIONS THAT UNDERSTAND THEM. |
| HUE028086T2 (en) | 2006-09-22 | 2016-11-28 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| DK2134374T3 (en) | 2007-03-14 | 2014-02-24 | Bionsil S R L In Liquidazione | BTK-INHIBITORS FOR USE IN PROCESSING chemotherapeutic agent RESISTANT TUMORS Epithelial |
| CA3001152A1 (en) | 2007-03-28 | 2008-10-09 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| WO2008144253A1 (en) | 2007-05-14 | 2008-11-27 | Irm Llc | Protein kinase inhibitors and methods for using thereof |
| MX2010000617A (en) | 2007-07-17 | 2010-05-17 | Plexxikon Inc | Compounds and methods for kinase modulation, and indications therefor. |
| US20090118276A1 (en) * | 2007-11-02 | 2009-05-07 | Wyeth | Thienopyrimidines, thienopyridines, and pyrrolopyrimidines as b-raf inhibitors |
| PE20091846A1 (en) | 2008-05-19 | 2009-12-16 | Plexxikon Inc | PIRROLO [2,3-d] -PYRIMIDINE DERIVATIVES AS KINE MODULATORS |
| WO2009143018A2 (en) | 2008-05-19 | 2009-11-26 | Plexxikon, Inc. | Compounds and methods for kinase modulation, and indications therefor |
| WO2010009342A2 (en) | 2008-07-16 | 2010-01-21 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase for the treatment of solid tumors |
| JP2012502104A (en) | 2008-09-10 | 2012-01-26 | カリプシス・インコーポレーテッド | Aminopyrimidine inhibitors against histamine receptors for the treatment of disease |
| WO2010036316A1 (en) | 2008-09-24 | 2010-04-01 | Yangbo Feng | Urea and carbamate compounds and analogs as kinase inhibitors |
| JP5731978B2 (en) | 2008-09-26 | 2015-06-10 | インテリカイン, エルエルシー | Heterocyclic kinase inhibitor |
| CL2009001884A1 (en) | 2008-10-02 | 2010-05-14 | Incyte Holdings Corp | Use of 3-cyclopentyl-3- [4- (7h-pyrrolo [2,3-d] pyrimidin-4-yl) -1h-pyrazol-1-yl) propanonitrile, janus kinase inhibitor, and use of a composition that understands it for the treatment of dry eye. |
| US20100261776A1 (en) | 2008-11-07 | 2010-10-14 | The Research Foundation Of State University Of New York | Bruton's tyrosine kinase as anti-cancer drug target |
| WO2010068806A1 (en) | 2008-12-10 | 2010-06-17 | Cgi Pharmaceuticals, Inc. | Amide derivatives as btk inhibitors in the treatment of allergic, autoimmune and inflammatory disorders as well as cancer |
| EP2416772A1 (en) | 2009-04-06 | 2012-02-15 | PTC Therapeutics, Inc. | Combinations of a hcv inhibitor such as bicyclic pyrrole derivatives and a therapeutic agent |
| WO2010117935A1 (en) | 2009-04-06 | 2010-10-14 | Schering Corporation | Compounds and methods for antiviral treatment |
| WO2010129053A2 (en) | 2009-05-05 | 2010-11-11 | Dana Farber Cancer Institute | Egfr inhibitors and methods of treating disorders |
| CA2771822C (en) | 2009-09-04 | 2020-08-11 | Daniel A. Erlanson | Bruton's tyrosine kinase inhibitors |
| WO2011029043A1 (en) | 2009-09-04 | 2011-03-10 | Biogen Idec Ma Inc. | Heteroaryl btk inhibitors |
| MX2012013196A (en) | 2010-05-12 | 2013-03-22 | Abbvie Inc | Pyrrolopyridine and pyrrolopyrimidine inhibitors of kinases. |
| US20120071497A1 (en) | 2010-06-03 | 2012-03-22 | Pharmacyclics, Inc. | Methods of treating abc-dlbcl using inhibitors of bruton's tyrosine kinase |
| WO2012058645A1 (en) | 2010-10-29 | 2012-05-03 | Biogen Idec Ma Inc. | Heterocyclic tyrosine kinase inhibitors |
| MX2014000130A (en) | 2011-06-28 | 2014-05-01 | Pharmacyclics Inc | Methods and compositions for inhibition of bone resorption. |
| KR20140058543A (en) | 2011-07-08 | 2014-05-14 | 노파르티스 아게 | Novel pyrrolo pyrimidine derivatives |
| EP3778896A1 (en) | 2011-08-09 | 2021-02-17 | Fred Hutchinson Cancer Research Center | Methods and compositions for inhibiting the growth and/or proliferation of myc-driven tumor cells |
| DE102011111400A1 (en) | 2011-08-23 | 2013-02-28 | Merck Patent Gmbh | Bicyclic heteroaromatic compounds |
| JP6506555B2 (en) | 2011-10-19 | 2019-04-24 | ファーマサイクリックス エルエルシー | Use of Breton-type tyrosine kinase (Btk) inhibitors |
| US9550781B2 (en) | 2011-11-14 | 2017-01-24 | Centaurus Biopharma Co., Ltd. | Kinase modulating compounds, compositions containing the same and use thereof |
-
2012
- 2012-07-07 KR KR1020147002986A patent/KR20140058543A/en not_active Application Discontinuation
- 2012-07-07 WO PCT/IB2012/001699 patent/WO2013008095A1/en active Application Filing
- 2012-07-07 JP JP2014517980A patent/JP6145451B2/en not_active Expired - Fee Related
- 2012-07-07 EA EA201490229A patent/EA201490229A1/en unknown
- 2012-07-07 BR BR112014000314A patent/BR112014000314A2/en not_active IP Right Cessation
- 2012-07-07 ES ES12761655.5T patent/ES2548414T3/en active Active
- 2012-07-07 AU AU2012282229A patent/AU2012282229B2/en not_active Expired - Fee Related
- 2012-07-07 US US14/130,536 patent/US9233111B2/en not_active Expired - Fee Related
- 2012-07-07 MX MX2014000338A patent/MX2014000338A/en unknown
- 2012-07-07 EP EP12761655.5A patent/EP2729466B1/en active Active
- 2012-07-07 CA CA2841111A patent/CA2841111A1/en not_active Abandoned
- 2012-07-07 CN CN201280033935.9A patent/CN103732596B/en active Active
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002038561A1 (en) | 2000-11-07 | 2002-05-16 | Novartis Ag | Indolylmaleimide derivatives as protein kinase c inhibitors |
| WO2003082859A1 (en) | 2002-04-03 | 2003-10-09 | Novartis Ag | Indolylmaleimide derivatives |
| WO2004052359A1 (en) | 2002-12-09 | 2004-06-24 | The Board Of Regents Of The University Of Texas System | Methods for selectively inhibiting janus tyrosine kinase 3 (jak3) |
| WO2004078163A2 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2005000833A1 (en) | 2003-05-19 | 2005-01-06 | Irm, Llc | Immunosuppressant compounds and compositions |
| WO2005066156A1 (en) | 2004-01-12 | 2005-07-21 | Cytopia Research Pty Ltd | Selective kinase inhibitors |
| WO2007090752A1 (en) | 2006-02-07 | 2007-08-16 | F. Hoffmann-La Roche Ag | Anthranilamide/2-amino-heteroarene carboxamide derivatives |
| WO2007145834A2 (en) | 2006-06-08 | 2007-12-21 | Amgen Inc. | Benzamide derivatives and uses related thereto |
| US20090105209A1 (en) | 2007-10-23 | 2009-04-23 | Roche Palo Alto Llc | BTK protein kinase inhibitors |
| WO2009077334A1 (en) | 2007-12-14 | 2009-06-25 | F. Hoffmann-La Roche Ag | Novel imidazo[1,2-a]pyridine and imidazo[1,2-b]pyridazine derivatives |
| US20090306041A1 (en) | 2008-02-05 | 2009-12-10 | Roche Palo Alto Llc | Inhibitors of Bruton's tyrosine kinase |
| WO2010000633A1 (en) | 2008-07-02 | 2010-01-07 | F. Hoffmann-La Roche Ag | Novel phenylpyrazinones as kinase inhibitors |
Non-Patent Citations (9)
| Title |
|---|
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY |
| "Remington's Pharmaceutical Sciences", 1990, MACK PRINTING COMPANY, pages: 1289 - 1329 |
| DAVIS,R.E. ET AL., NATURE, vol. 463, 2010, pages 88 - 92 |
| FATIH M UCKUN ET AL: "Bruton's tyrosine kinase as a molecular target in treatment of leukemias and lymphomas as well as inflammatory disorders and autoimmunity", EXPERT OPINION ON THERAPEUTIC PATENTS, INFORMA HEALTHCARE, GB, vol. 20, no. 11, 1 November 2010 (2010-11-01), pages 1457 - 1470, XP008157228, ISSN: 1354-3776, DOI: 10.1517/13543776.2010.517750 * |
| HATA,D. ET AL., J. EXP. MED., vol. 187, 1998, pages 1235 - 1247 |
| J. MED. CHEM., vol. 52, no. 14, 2009, pages 4329 - 4337 |
| J. MED. CHEM., vol. 52, no. 19, 2009, pages 5950 - 5966 |
| STAHL; WERMUTH: "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", 2002, WILEY-VCH |
| STEINBERG,B.J. ET AL., J. CLIN. INVEST, vol. 70, 1982, pages 587 - 597 |
Cited By (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9233111B2 (en) | 2011-07-08 | 2016-01-12 | Novartis Ag | Pyrrolo pyrimidine derivatives |
| WO2013157021A1 (en) | 2012-04-20 | 2013-10-24 | Advinus Therapeutics Limited | Bicyclic compounds, compositions and medicinal applications thereof |
| US9233983B2 (en) | 2012-04-20 | 2016-01-12 | Advinus Therapeutics Limited | Substituted hetero-bicyclic compounds, compositions and medicinal applications thereof |
| AU2013250726B2 (en) * | 2012-04-20 | 2017-01-05 | Advinus Therapeutics Limited | Substituted hetero-bicyclic compounds, compositions and medicinal applications thereof |
| WO2013157022A1 (en) | 2012-04-20 | 2013-10-24 | Advinus Therapeutics Limited | Substituted hetero-bicyclic compounds, compositions and medicinal applications thereof |
| EP3550031A1 (en) | 2012-07-24 | 2019-10-09 | Pharmacyclics, LLC | Mutations associated with resistance to inhibitors of bruton's tyrosine kinase (btk) |
| US9512084B2 (en) | 2013-11-29 | 2016-12-06 | Novartis Ag | Amino pyrimidine derivatives |
| US11180460B2 (en) | 2013-11-29 | 2021-11-23 | Novartis Ag | Amino pyrimidine derivatives |
| US11673868B2 (en) | 2013-11-29 | 2023-06-13 | Novartis Ag | Amino pyrimidine derivatives |
| US10457647B2 (en) | 2013-11-29 | 2019-10-29 | Novartis Ag | Amino pyrimidine derivatives |
| JP2020105181A (en) * | 2013-12-02 | 2020-07-09 | ファーマサイクリックス エルエルシー | Methods of treating and preventing alloantibody driven chronic graft versus host disease |
| US11623921B2 (en) | 2014-10-24 | 2023-04-11 | Bristol-Myers Squibb Company | Indole carboxamide compounds |
| EP3042903A1 (en) | 2015-01-06 | 2016-07-13 | Advinus Therapeutics Limited | Substituted hetero-bicyclic compounds, compositions and medicinal applications thereof |
| US11407749B2 (en) | 2015-10-19 | 2022-08-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11572366B2 (en) | 2015-11-19 | 2023-02-07 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2017106634A1 (en) * | 2015-12-17 | 2017-06-22 | Incyte Corporation | N-phenyl-pyridine-2-carboxamide derivatives and their use as pd-1/pd-l1 protein/protein interaction modulators |
| US11866435B2 (en) | 2015-12-22 | 2024-01-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11535615B2 (en) | 2015-12-22 | 2022-12-27 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11608337B2 (en) | 2016-05-06 | 2023-03-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11673883B2 (en) | 2016-05-26 | 2023-06-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11873309B2 (en) | 2016-06-20 | 2024-01-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11718605B2 (en) | 2016-07-14 | 2023-08-08 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11613536B2 (en) | 2016-08-29 | 2023-03-28 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2018053437A1 (en) | 2016-09-19 | 2018-03-22 | Mei Pharma, Inc. | Combination therapy |
| US11339149B2 (en) | 2016-12-22 | 2022-05-24 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10308644B2 (en) | 2016-12-22 | 2019-06-04 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11787793B2 (en) | 2016-12-22 | 2023-10-17 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11465981B2 (en) | 2016-12-22 | 2022-10-11 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10806785B2 (en) | 2016-12-22 | 2020-10-20 | Incyte Corporation | Immunomodulator compounds and methods of use |
| US10793565B2 (en) | 2016-12-22 | 2020-10-06 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11566026B2 (en) | 2016-12-22 | 2023-01-31 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10800768B2 (en) | 2016-12-22 | 2020-10-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11608334B2 (en) | 2017-02-08 | 2023-03-21 | The National Institutes of Pharmaceutical R&D Co., Ltd. | Pyrrolo-aromatic heterocyclic compound, preparation method therefor, and medical use thereof |
| EP3556761A4 (en) * | 2017-02-08 | 2019-12-25 | The National Institutes Of Pharmaceutical Research | Pyrrolo-aromatic heterocyclic compound, preparation method therefor, and medical use thereof |
| US11613543B2 (en) | 2018-03-26 | 2023-03-28 | Novartis Ag | Substituted pyrrolo[2,3-d]pyrimidines as Bruton's Tyrosine Kinase inhibitors |
| EP4166557A1 (en) | 2018-03-26 | 2023-04-19 | Novartis AG | Intermediates of n-(3-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)benzamide derivatives |
| WO2019186343A1 (en) | 2018-03-26 | 2019-10-03 | Novartis Ag | N-(3-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)benzamide derivatives |
| WO2019186358A1 (en) | 2018-03-26 | 2019-10-03 | Novartis Ag | 3-hydroxy-n-(3-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)pyrrolidine-1-carboxamide derivatives |
| US11541056B2 (en) | 2018-03-26 | 2023-01-03 | Novartis Ag | 3-hydroxy-N-(3-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)pyrrolidine-1-carboxamide derivatives |
| US10669271B2 (en) | 2018-03-30 | 2020-06-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11124511B2 (en) | 2018-03-30 | 2021-09-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11414433B2 (en) | 2018-05-11 | 2022-08-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10618916B2 (en) | 2018-05-11 | 2020-04-14 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10906920B2 (en) | 2018-05-11 | 2021-02-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11753406B2 (en) | 2019-08-09 | 2023-09-12 | Incyte Corporation | Salts of a PD-1/PD-L1 inhibitor |
| US11401279B2 (en) | 2019-09-30 | 2022-08-02 | Incyte Corporation | Pyrido[3,2-d]pyrimidine compounds as immunomodulators |
| US11866451B2 (en) | 2019-11-11 | 2024-01-09 | Incyte Corporation | Salts and crystalline forms of a PD-1/PD-L1 inhibitor |
| US11780836B2 (en) | 2020-11-06 | 2023-10-10 | Incyte Corporation | Process of preparing a PD-1/PD-L1 inhibitor |
| US11760756B2 (en) | 2020-11-06 | 2023-09-19 | Incyte Corporation | Crystalline form of a PD-1/PD-L1 inhibitor |
| US11866434B2 (en) | 2020-11-06 | 2024-01-09 | Incyte Corporation | Process for making a PD-1/PD-L1 inhibitor and salts and crystalline forms thereof |
| WO2022189859A1 (en) | 2021-03-12 | 2022-09-15 | Novartis Ag | Fatty acid-bifunctional degrader conjugates and their methods of use |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2012282229B2 (en) | 2015-05-07 |
| EP2729466B1 (en) | 2015-08-19 |
| EA201490229A1 (en) | 2014-05-30 |
| US20140243306A1 (en) | 2014-08-28 |
| CN103732596B (en) | 2016-06-01 |
| AU2012282229A1 (en) | 2014-01-16 |
| ES2548414T3 (en) | 2015-10-16 |
| CN103732596A (en) | 2014-04-16 |
| BR112014000314A2 (en) | 2017-01-10 |
| CA2841111A1 (en) | 2013-01-17 |
| US9233111B2 (en) | 2016-01-12 |
| JP6145451B2 (en) | 2017-06-14 |
| EP2729466A1 (en) | 2014-05-14 |
| KR20140058543A (en) | 2014-05-14 |
| JP2014520793A (en) | 2014-08-25 |
| MX2014000338A (en) | 2014-05-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2729466B1 (en) | Novel pyrrolo pyrimidine derivatives | |
| US11673868B2 (en) | Amino pyrimidine derivatives | |
| EP2897961B1 (en) | Dihydropyrrolidino-pyrimidines as kinase inhibitors | |
| US20150216867A1 (en) | Compounds for use in gastric complication | |
| TW201425313A (en) | Novel pyrrolo pyrimidine derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12761655 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012761655 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2014517980 Country of ref document: JP Kind code of ref document: A Ref document number: 2841111 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2014/000338 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2012282229 Country of ref document: AU Date of ref document: 20120707 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20147002986 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201490229 Country of ref document: EA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014000314 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14130536 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 112014000314 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140107 |