WO2012138648A1 - Compositions and methods for modulating lpa receptors - Google Patents
Compositions and methods for modulating lpa receptors Download PDFInfo
- Publication number
- WO2012138648A1 WO2012138648A1 PCT/US2012/031985 US2012031985W WO2012138648A1 WO 2012138648 A1 WO2012138648 A1 WO 2012138648A1 US 2012031985 W US2012031985 W US 2012031985W WO 2012138648 A1 WO2012138648 A1 WO 2012138648A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- methyl
- pyrazol
- amino
- carbonyl
- Prior art date
Links
- 0 C*=C(C=CC(C)=C)c1c(NC(O*c2ccccc2)=O)[n](*)nc1 Chemical compound C*=C(C=CC(C)=C)c1c(NC(O*c2ccccc2)=O)[n](*)nc1 0.000 description 8
- KITYDWVANPQFIU-UHFFFAOYSA-N CC(C)(C(C)(C)OBc(ccc(-c1ccc(C2(CC2)C(OC)=O)cn1)c1)c1OC)O Chemical compound CC(C)(C(C)(C)OBc(ccc(-c1ccc(C2(CC2)C(OC)=O)cn1)c1)c1OC)O KITYDWVANPQFIU-UHFFFAOYSA-N 0.000 description 1
- KZWNEHHPHBUBGM-UHFFFAOYSA-N CC(N(CC1)CCC1(C(O)=O)c(cc1)ccc1Br)=O Chemical compound CC(N(CC1)CCC1(C(O)=O)c(cc1)ccc1Br)=O KZWNEHHPHBUBGM-UHFFFAOYSA-N 0.000 description 1
- IPWKHHSGDUIRAH-UHFFFAOYSA-N CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C Chemical compound CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 1
- VAWYJERIKSKEPK-UHFFFAOYSA-N CC1(C)OB(c2ccc(CCC(OC)=O)cc2)OC1(C)C Chemical compound CC1(C)OB(c2ccc(CCC(OC)=O)cc2)OC1(C)C VAWYJERIKSKEPK-UHFFFAOYSA-N 0.000 description 1
- IKIULKMEJZZLFL-GFCCVEGCSA-N C[C@H](c1ccccc1)OC(Nc([n](C)nc1)c1-c(nc1)ccc1Br)=O Chemical compound C[C@H](c1ccccc1)OC(Nc([n](C)nc1)c1-c(nc1)ccc1Br)=O IKIULKMEJZZLFL-GFCCVEGCSA-N 0.000 description 1
- GPWMPBYYSVXMDN-FMKOIGJVSA-O C[C@H](c1ccccc1Cl)OC(N/C(/NC)=C(/C=[NH2+])\c(cc1)ccc1-c1ccc(CCC(OC)=O)cc1)=O Chemical compound C[C@H](c1ccccc1Cl)OC(N/C(/NC)=C(/C=[NH2+])\c(cc1)ccc1-c1ccc(CCC(OC)=O)cc1)=O GPWMPBYYSVXMDN-FMKOIGJVSA-O 0.000 description 1
- RGASNHQOGFREGG-CYBMUJFWSA-N C[C@H](c1ccccc1Cl)OC(Nc([n](C)cc1)c1-c(cc1)ccc1Br)=O Chemical compound C[C@H](c1ccccc1Cl)OC(Nc([n](C)cc1)c1-c(cc1)ccc1Br)=O RGASNHQOGFREGG-CYBMUJFWSA-N 0.000 description 1
- LWELFBDXWZZCNO-GOSISDBHSA-N C[C@H](c1ccccc1Cl)OC(Nc([n](C)nc1)c1-c(cc1)ccc1-c1ccc(CCC(O)=O)cc1)=O Chemical compound C[C@H](c1ccccc1Cl)OC(Nc([n](C)nc1)c1-c(cc1)ccc1-c1ccc(CCC(O)=O)cc1)=O LWELFBDXWZZCNO-GOSISDBHSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D231/38—Nitrogen atoms
- C07D231/40—Acylated on said nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/04—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles
- C07D249/06—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles with aryl radicals directly attached to ring atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/10—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the invention provides a compound having Formula (2):
- R 1 and R 2 are independently hydrogen or fluoro.
- R 1 and R 2 in each -(CR ⁇ 2 )- repeating group are defined independently, and encompass identical and non-identical -(CR ⁇ 2 )- repeating groups.
- R 3 , R 4a , R 4b , R 7 , R 8 , m, n and p are as defined in Formula (1); or
- the invention provides a compound of Formula (4):
- the invention provides a compound selected from the group consisting of:
- the invention provides a compound selected from the group consisting of:
- the invention provides a method for treating a LPA-dependent or LPA-mediated disease or condition comprising administering to a subject in need thereof a therapeutically effective amount of a compound having Formula (1), (2), (3) or (4) or a pharmaceutically acceptable salt thereof, and optionally in combination with a second therapeutic agent.
- the invention also provides a compound having Formula (1), (2), (3) or (4) or a pharmaceutically acceptable salt thereof, and optionally in combination with a second therapeutic agent, for use in the treatment of a LPA-dependent or LPA-mediated disease or condition.
- the invention further provides the use of a compound having Formula (1), (2), (3) or
- the second therapeutic agent can be selected from corticosteroids, immunosuppresant, analgesics, anti-cancer agent, anti-inflammatories, chemokine receptor antagonists, bronchodilators, leukotriene receptor antagonists, leukotriene formation inhibitors, monoacylglycerol kinase inhibitors, phospholipase A] inhibitors, phospholipase A 2 inhibitors, and lysophospholipase D (lysoPLD) inhibitors, autotaxin inhibitors, decongestants,
- halogen refers to fluoro, chloro, bromo, and iodo; and more particularly, fluoro or chloro.
- stereoisomer refers to a compound made up of the same atoms bonded by the same bonds but having different three-dimensional structures, which are not interchangeable.
- the present invention contemplates various stereoisomers and mixtures thereof and includes “enantiomers”, which refers to two stereoisomers whose molecules are non- superimposeable mirror images of one another.
- a subject is "in need of a treatment if such subject would benefit biologically, medically or in quality of life from such treatment.
- Figure 1 depicts the X-ray powder diffraction pattern of (R)-l-(4'-(l-methyl-5-((l- phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid (6a) and (R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid potassium salt (6b).
- the present invention relates to compositions and methods for modulating the activity of LPA receptors.
- the present invention relates to compounds which act as inhibitors of LPA, and methods of using such compounds to treat a disease or condition associated with one or more LPA receptors.
- Various embodiments of the invention are described herein. It will be recognized that features specified in each embodiment may be combined with other specified features to provide further embodiments.
- nyl, phenyl fused to a cyclopentyl, C3-7 cycloalkyl, spiro[2,5]octanyl, or C3 of the pyridyl ring is attached to -(CR 1 R 2 ) P -R 3 ;
- Z 1 and Z 2 are independently CH or N; or Z 2 is C if attached to R 4a ;
- R 3 is -C0 2 H, -C(0)NR-(CRR')o-2-CN, -C(0)NRS0 2 R 10 or -NR-C(0)R 10 ;
- R 4a and R 4b are independently hydrogen, halo, Ci_ 6 alkyl, Ci_ 6 alkoxy or Ci_ 6 hydroxyalkyl;
- R 7 and R 8 are independently hydrogen, Ci ⁇ alkyl or halo
- R 9 is -C(0)R n , -C(0)OR n or S0 2 R n ;
- R and R' are independently hydrogen or Ci_6 alkyl
- n and n are independently 1-4;
- p is 0-2;
- the invention provides a compound having Formula (3):
- R 1 and R 2 are independently hydrogen or fluoro;
- R 1 and R 2 together form cyclopropyl
- R 3 , R 4a , R 4b , R 7 , R 8 , m, n and p are as defined in Formula (1); or
- the invention provides a compound having Formula (1), provided said compound is not a compound of Formula (3).
- the invention provides a compound of Formula (4):
- the invention provides a compound having Formula (5) or (5 A): wherein B is phenyl, C3-7 cycloalkyl, a 4-6 membered heterocycle comprising 1-2 heteroatoms selected from N, NR 9 , O and S(0) q ; N wherein CI of the pyridyl ring is attached to -(CR 1 R 2 ) P -R 3 , a 5-6 membered heteroaryl comprising 2-3 heteroatoms selected from N, O and S; or a 9-10 membered bicyclic carbocyclic or bicyclic heterocyclic ring comprising 1-3 heteroatoms selected from N, NR 9 , O and S;
- R 2 is hydrogen, Ci_6 alkyl or halo
- R 3 is -CO 2 R, cyano, -C(0)NRS0 2 R 10 , -S0 2 -NR-C(0)R 10 , -NR-C(0)R 10 , -NR-SO 2 R 10 or tetrazolyl;
- R 4a and R 4b are independently halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONR 2 or C0 2 R;
- R 5 is methyl or ethyl
- R 6 is Ci_6 alkyl, Ci_ 6 haloalkyl or -(CR 2 )-0(Ci_ 4 alkyl);
- R 6b is Ci_6 alkyl; C3_7 cycloalkyl that is monocyclic or optionally fused to a phenyl that is unsubstituted or substituted with R 7 and R 8 ; phenyl substituted with R 7 and R 8 ; a 5-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O or S, and unsubstituted or substituted by 1-2 R 9 ; or a 5-6 membered heteroaryl comprising 1-3 heteroatoms selected from N, O and S, and unsubstituted or substituted by 1-2 R 9 ;
- R 7 and R 8 are independently hydrogen, methyl, methoxy or halo
- R 9 is (Ci_6 alkyl), -C(0)R n , -C(0)OR n , -C(0)NR n R 12 or S0 2 R n ;
- R 10 , R 11 and R 12 are independently Ci ⁇ alkyl, haloCi_ 4 alkyl, cyclopropyl, phenyl unsubstituted or substituted by Ci_ 4 alkyl;
- n, p and q are independently 0-2; or
- R 1 , R 2 , R 3 , R 4a , R 4b , R 5 , R 6 , R 7 , R 8 , m, n and p are as defined in Formula (5); or a stereoisomer, prodrug or pharmaceutically acceptable salt thereof.
- the invention provides a compound of Formula (5), (5 A) or (6), wherein a substituent is defined independently, collectively, or in any combination or subcombination, as follows:
- B is phenyl, C3-7 cycloalkyl, a 4-6 membered heterocycle comprising 1-2 heteroatoms selected from N, NR 9 , O and S(0) q ; wherein CI of the pyridyl ring is attached to -(CR 1 R 2 ) P -R 3 , a 5-6 membered heteroaryl comprising 2-3 heteroatoms selected from N, O and S; or a 9-10 membered bicyclic carbocyclic or bicyclic heterocyclic ring comprising 1-3 heteroatoms selected from N, NR 9 , O and S; and particularly, wherein B phenyl, pyrazolyl, cyclohexyl, piperazinyl, 2-oxo- 1 ,2-dihydropyridin-4-yl, cyclopropyl, cyclobutyl, cyclopentyl, azetidinyl, pyrrolidinyl, N wherein CI of the pyridyl
- tetrahydroisoquinolinyl wherein the -NH- moiety in said azetidinyl, pyrrolidinyl or tetrahydroisoquinolinyl is optionally substituted by -C(0)R n or S0 2 R n and R 11 is C 1-4 alkyl (e.g., N-acetyl-pyrrolidinyl, N-methylsulfonyl-pyrrolidinyl, N-acetyl-tetrahydroisoquinolinyl and the like); and more particularly, wherein B is phenyl, pyrazolyl, N wherein CI of the pyridyl ring is attached to -(CR 1 R 2 ) P -R 3 , cyclohexyl, piperazinyl or 2-oxo- 1,2- dihydropyridin-4-yl ;
- R 1 and R 2 together form cyclopropyl, cyclobutyl, or a 3-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O and S, and wherein said cyclopropyl, cyclobutyl, or 3-6 membered heterocycle is optionally substituted by 1-2 R 9 ; and particularly, wherein R 1 and R 2 together form cyclopropyl, oxetanyl, or azetidinyl wherein the hydrogen in NH is optionally replaced by -C(0)R n or S0 2 R n and R 11 and R 12 are independently Ci_ 4 alkyl (e.g., N-acetyl-azetidinyl or N-methylsulfonyl-azetidinyl);
- R 3 is -C0 2 R, cyano, -C(0)NRS0 2 R 10 , -S0 2 -NR-C(0)R 10 , -NR-C(0)R 10 , -NR-
- R 3 is-C0 2 R and R is hydrogen or Ci_6 alkyl; and more particularly, wherein R 3 is-C0 2 R and R is hydrogen;
- R 4a and R 4b if present are independently halogen, Ci_ 6 alkyl, Ci_ 6 alkoxy, haloCi_ 6 alkoxy, cyano, CONR 2 or C0 2 R; and particularly, wherein R 4a and R 4b if present, are independently halo or Ci_6 alkyl (particularly, fluoro, chloro, or methyl); and more particularly, wherein R 4a if present is methyl or fluoro, and R 4b if present is fluoro;
- R 5 is methyl or ethyl; and more particularly, wherein R 5 is methyl;
- R 6 is Ci_6 alkyl, Ci_ 6 haloalkyl or -(CR 2 )-0(C 1-4 alkyl); and particularly, wherein R 6 is methyl, ethyl, propyl, t-butyl or methoxy methylene;
- R 6a is hydro gen or Ci_6 alkyl (particularly methyl); and particularly, wherein R a is hydrogen;
- R 6b is Ci-6 alkyl; C3-7 cycloalkyl that is monocyclic or optionally fused to a phenyl that is unsubstituted or substituted with R 7 and R 8 (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, dihydroindenyl); phenyl substituted with R 7 and R 8 ; a 5-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O or S, and unsubstituted or substituted by 1-2 R 9 (e.g., tetrahydrofuranyl, tetrahydropyranyl, piperidyl, N-Boc piperidyl); or a 5-6 membered heteroaryl comprising 1-3 heteroatoms selected from N, O and S, and unsubstituted or substituted by 1-2 R 9 (e.g., pyridyl, oxazolyl,
- R 9 is (Ci_6 alkyl), -C(0)R n , -C(0)OR n , -C(0)NR n R 12 or S0 2 R n ;
- R 10 , R 11 and R 12 are independently Ci ⁇ alkyl, haloCi ⁇ alkyl, cyclopropyl, phenyl unsubstituted or substituted by Ci_ 4 alkyl;
- n) R is hydrogen or Ci_6 alkyl
- n 0-2; and more particularly, wherein m and n are 0-1 ;
- p) p and q are independently 0-2; or
- the invention provides a compound of Formula (7),
- n are independently 0- 1 ;
- R 3 , R 4a , R 4b , R 5 , R 6 , R 7 and R 8 are as defined in Formula (1);
- the invention provides a compound of Formula (7),
- n are independently 0- 1 ;
- R 3 , R 4a , R 4b , R 5 , R 6 , R 7 and R 8 are as defined independently, collectively, or in any combination or sub-combination as follows:
- R 3 is -C0 2 R, cyano, -C(0)NRS0 2 R 10 , -S0 2 -NR-C(0)R 10 , -NR-C(0)R 10 , -NR- SO 2 R 10 or tetrazolyl; and particularly, wherein R 3 is-C0 2 R and R is hydrogen or Ci_ 6 alkyl; and more particularly, wherein R 3 is-C0 2 R and R is hydrogen;
- R 4a and R 4b if present are independently halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONR 2 or C0 2 R; and particularly, wherein R 4a and R 4b if present, are independently halo or Ci_6 alkyl (particularly, fluoro, chloro, or methyl); and more particularly, wherein R 4a if present is methyl or fluoro, and R 4b if present is fluoro;
- R 5 is methyl or ethyl; and more particularly, wherein R 5 is methyl;
- R 6 is Ci-6 alkyl, Ci_6 haloalkyl or alkyl ); and particularly, wherein R 6 is methyl, ethyl, propyl, t-butyl or methoxymethylene;
- R 7 and R 8 are independently hydrogen, methyl, methoxy or halo; and particularly, wherein R 7 and R 8 are independently hydrogen, methyl or halo; and more particularly, wherein R 7 is hydrogen and chloro, and R 8 is hydrogen or fluoro;
- R 10 is Ci-4 alkyl, haloCi ⁇ alkyl, cyclopropyl, phenyl unsubstituted or substituted by alkyl;
- R is hydrogen or Ci_6 alkyl; or a stereoisomer, prodrug or pharmaceutically acceptable salt thereof.
- the invention provides a compound of Formula (5A), wherein Z is N; and B, R 1 , R 2 , R 3 , R 4a , R 4b , R 5 , R 6 , R 6a , R 6b , m and n are as defined
- the invention provides a compound of Formula (5 A), wherein:
- B is phenyl
- R 1 and R 2 together form cyclopropyl
- R 3 is -C0 2 R
- R 6b is phenyl substituted with R 7 and R 8 ;
- R 7 and R 8 are independently hydrogen or halo
- R is H
- R 4a , R 4b , R 5 , R 6 , R 6a , m and n are as defined in Formula (1).
- the invention provides a compound of Formula (5), (5 A) or (6), wherein R 1 and R 2 are independently hydrogen or fluoro.
- the invention provides a compound having Formula (5'), (5B) or (5C):
- A is phenyl or a 5-6 membered heteroaryl comprisingl-3 heteroatoms selected from N, O and S;
- B is phenyl, C3-7 cycloalkyl, a 4-7 membered heterocycle comprising 1-2 heteroatoms selected from N, NR 9 , O and S(0) q ; a 5-6 membered heteroaryl comprising 1-3 heteroatoms selected from N, O and S; or a 9-12 membered bicyclic carbocyclic or bicyclic heterocyclic ring comprising 1-3 heteroatoms selected from N, NR 9 , O and S;
- L 1 and L 2 are independently a bond or (CR 2 );
- R 1 is hydrogen, Ci_ 6 alkyl, Ci_3 alkoxy, halo or C0 2 R;
- R 2 is hydrogen, Ci_6 alkyl or halo
- R 1 and R 2 together form cyclopropyl, cyclobutyl, or a 3-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O and S, and wherein said cyclopropyl, cyclobutyl, or 3-6 membered heterocycle is optionally substituted by 1-2 R 9 ;
- R 3 is -C0 2 R, cyano, -C(0)NRS0 2 R 10 , S0 2 NR 2 , -S0 2 -NR-C(0)R 10 , -NR-C(0)R 10 , -NR- SO 2 R 10 or tetrazolyl;
- R 4a and R 4b are independently halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONR 2 or C0 2 R;
- R 5 is methyl or ethyl
- R 6 is Ci_6 alkyl, Ci_ 6 haloalkyl or -(CR 2 )-0(Ci_ 4 alkyl);
- R 6a is hydrogen or Ci_6 alkyl
- R 6b is Ci-6 alkyl; C3-7 cycloalkyl that is monocyclic or optionally fused to a phenyl that is unsubstituted or substituted with R 7 and R 8 ; phenyl substituted with R 7 and R 8 ; a 5-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O or S, and unsubstituted or substituted by 1-2 R 9 ; or a 5-6 membered heteroaryl comprising 1-3 heteroatoms selected from N, O and S, and unsubstituted or substituted by 1-2 R 9 ;
- R 7 and R 8 are independently hydrogen, methyl, methoxy or halo;
- R 9 is (Ci_6 alkyl), -C(0)R n , -C(0)OR n , -C(0)NR n R 12 or S0 2 R n ;
- R 10 , R 11 and R 12 are independently Ci ⁇ alkyl, haloCi-4 alkyl, cyclopropyl, phenyl unsubstituted or substituted by Ci_ 4 alkyl;
- R is hydrogen or Ci_ 6 alkyl
- k 0-1
- n, p and q are independently 0-2; or
- the invention provides a compound of Formula (5'), (5B) or (5C), wherein a substituent is defined independently, collectively, or in any combination or sub-combination, as follows:
- B is phenyl, C3-7 cycloalkyl, a 4-6 membered heterocycle comprising 1-2 heteroatoms selected from N, NR 9 , O and S(0) q ; wherein CI of the pyridyl ring is attached to a 5-6 membered heteroaryl comprising 2-3 heteroatoms selected from N, O and S; or a 9-10 membered bicyclic carbocyclic or bicyclic heterocyclic ring comprising 1-3 heteroatoms selected from N, NR 9 , O and S; and particularly, wherein B is phenyl, pyrazolyl, cyclohexyl, piperazinyl, 2-oxo-l,2-dihydropyridin-
- R 2 is hydrogen, Ci_ 6 alkyl or halo; and particularly, wherein R 2 is hydrogen or halo (fluoro); or d) R 1 and R 2 together form cyclopropyl, cyclobutyl, or a 3-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O and S, and wherein said cyclopropyl, cyclobutyl, or 3-6 membered heterocycle is optionally substituted by 1-2 R 9 ; and particularly, wherein R 1 and R 2 together form cyclopropyl, oxetanyl, or azetidinyl wherein the hydrogen in NH is optionally replaced by -C(0)R n or S0 2 R n and R 11 and R 12 are independently Ci_ 4 alkyl (e.g., N-acetyl-azetidinyl or N-methylsulfonyl-azetidinyl);
- R 3 is -C0 2 R, cyano, -C(0)NRS0 2 R 10 , -S0 2 -NR-C(0)R 10 , -NR-C(0)R 10 , -NR- SO 2 R 10 or tetrazolyl; and particularly, wherein R 3 is-C0 2 R and R is hydrogen or Ci_6 alkyl; and more particularly, wherein R 3 is-C0 2 R and R is hydrogen;
- R 4a and R 4b if present are independently halogen, Ci_6 alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONR 2 or C0 2 R; and particularly, wherein R 4a and R 4b if present, are independently halo, Ci_6 alkyl or haloCi_6 alkyl (particularly, fluoro, chloro, methyl or trifluoromethyl); and more particularly, wherein R 4a if present is methyl or fluoro, and R 4b if present is fluoro;
- R 5 is methyl or ethyl; and more particularly, wherein R 5 is methyl;
- R 6 is Ci-6 alkyl, Ci_6 haloalkyl or alkyl); and particularly, wherein R 6 is methyl, ethyl, propyl, t-butyl or methoxy methylene;
- R 6a is hydro gen or Ci_ 6 alkyl (particularly methyl); and particularly, wherein R a is hydrogen;
- R 6b is Ci-6 alkyl; C3-7 cycloalkyl that is monocyclic or optionally fused to a phenyl that is unsubstituted or substituted with R 7 and R 8 (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, dihydroindenyl); phenyl substituted with R 7 and R 8 ; a 5-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O or S, and unsubstituted or substituted by 1-2 R 9 (e.g., tetrahydrofuranyl, tetrahydropyranyl, piperidyl, N-Boc piperidyl); or a 5-6 membered heteroaryl comprising 1-3 heteroatoms selected from N, O and S, and unsubstituted or substituted by 1-2 R 9 (e.g., pyridyl, oxazolyl,
- R 9 is (Ci_6 alkyl), -C(0)R n , -C(0)OR n , -C(0)NR n R 12 or S0 2 R n ;
- R 10 , R 11 and R 12 are independently Ci ⁇ alkyl, haloCi ⁇ alkyl, cyclopropyl, phenyl unsubstituted or substituted by C 1-4 alkyl;
- n) R is hydrogen or Ci_6 alkyl
- n 0-2; and more particularly, wherein m and n are 0-1 ;
- the invention provides a compound having Formula (8):
- A is phenyl or 5-6 membered heteroaryl containing 1-3 heteroatoms selected from N, O and S;
- R 1 is Ci-6 alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, hydroxy, hydroxyCi-6 alkyl, halogen, cyano, acetonitrileoxy, NR 2 , C0 2 R, OS0 2 R 10 , S0 2 NR-L 1 -R 9 ,-L-NRC(0)R 10 , -L- NRS0 2 R 9 , -L-NRC0 2 R 10 , L-NRSOz-I ⁇ -COzR or -NRS0 2 NR 2 ; or R 1 is aryl, C 5 _ 7 cycloalkyl, or 4-6 membered heterocyclyl containing 1-3 heteroatoms selected from N, O and S, , each of which is optionally substituted with -L-C0 2 R or -L-tetrazolyl;
- R 2 is hydrogen, halogen or Ci_6 alkoxy
- R 1 and R 2 together with Ring A form a bicyclic 9-10 membered aryl or heteroaryl, said heteroaryl containing 1-3 heteroatoms selected from N, O and S;
- R 3 is halogen, Ci_6 alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONRR 9 or
- R 4 is hydrogen, cyclopropyl-Ci_ 2 alkyl or Ci_6 alkyl optionally substituted with 1-3 groups selected from cyano, halogen, hydroxy, Ci_6 alkoxy, C0 2 R or NR 2 ;
- R 5 and R 6 are independently hydrogen, Ci_6 alkyl or haloCi_6 alkyl; or R 5 and R 6 together with the carbon atoms to which they are attached form a C3-7 cycloalkyl or 5-6 membered heterocyclyl containing 1-2 heteroatoms selected from N, O and S;
- R 7 is phenyl, 5-6 membered heteroaryl containing 1-3 heteroatoms selected from N, O and S; 5-6 membered heterocyclyl containing 1-2 heteroatoms selected from N, O and S; or a bicyclic 9-10 membered aryl or heteroaryl, said heteroaryl containing 1-3 heteroatoms selected from N, O and S; wherein R 7 is optionally substituted with R 11 and 1-2 R 12 ;
- R 8 is hydrogen, halogen, cyano, Ci_ 6 alkyl, haloCi_ 6 alkyl, Ci_ 6 alkoxy, haloCi_ 6 alkoxy, C(0)NRR 10 or C0 2 R 10 ;
- R 9 is Ci-6 alkyl, haloCi_6 alkyl, C3-7 cycloalkyl or phenyl; wherein said C3-7 cycloalkyl, phenyl is optionally substituted with Ci_6 alkyl, C0 2 R or tetrazolyl;
- R 10 is Ci_6 alkyl or haloCi_ 6 alkyl
- R 11 is halogen or Ci_6 alkyl
- R 12 is halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkyl, haloCi_6 alkoxy or cyano;
- R is H or Ci-6 alkyl
- R and R 9 together with the nitrogen atom in S0 2 NRR 9 , or R and R 10 together with the nitrogen atom in C(0)NRR 10 form a 5-6 membered heterocyclic ring;
- L and L 1 are independently a bond or (CR a R a ) k ;
- R a is H, Ci-6 alkyl, hydroxy, halo or C1-3 alkoxy;
- k 0-4;
- n 0-2;
- the invention provides a compound having Formula
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 and m are as defined in Formula (8); or a stereoisomer or pharmaceutically acceptable salt thereof.
- the invention provides a compound having Formula (10
- R 11 is halogen or Ci_6 alkyl
- R 12 is halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkyl, haloCi_6 alkoxy or cyano;
- n 0-2;
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 and R 8 are as defined in Formula (8); or
- the invention provides a compound of Formula (8), (9A), (9B), (9C), (9D), (10A) or (10B), wherein a substituent is defined independently, collectively, or in any combination or sub-combination, as follows:
- A is phenyl or 5-6 membered heteroaryl containing 1-3 heteroatoms selected from N, O and S; and particularly, wherein A is phenyl, pyridyl or thiazolyl;
- R 1 is Ci-6 alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, hydroxy, hydroxyCi-6 alkyl, halogen, cyano, acetonitrileoxy, NR 2 , C0 2 R, OS0 2 R 10 , S0 2 NR-L 1 -R 9 ,-L-NRC(0)R 10 , -L- NRS0 2 R 9 , -L-NRC0 2 R 10 , L-NRS0 2 -L 1 -C0 2 R or -NRS0 2 NR 2 ; or R 1 is aryl, C 5 - 7 cycloalkyl, or 4-6 membered heterocyclyl containing 1-3 heteroatoms selected from N, O and S, , each of which is optionally substituted with -L-C0 2 R or -L-tetrazolyl; and particularly, wherein R 1 is Ci-6 alkyl, halo
- R 9 is Ci-6 alkyl, haloCi_6 alkyl, cyclopropyl, phenyl optionally substituted with Ci_6 alkyl;
- R is Ci-6 alkyl or haloCi-6 alkyl;
- L and L 1 are independently a bond or (CR a R a ) k and k is 0-2;
- R and R a are independently H or Ci_ 6 alkyl
- R 2 is hydrogen, halogen or Ci_ 6 alkoxy; and particularly, wherein R 2 is hydrogen, methoxy or fluoro;
- R 1 and R 2 together with Ring A form a bicyclic 9-10 membered aryl or heteroaryl, said heteroaryl containing 1-3 heteroatoms selected from N, O and S; and particularly, wherein R 1 and R 2 together with Ring A form an indole, indazole, benzimidazole or benzotriazolyl;
- R 3 is halogen, Ci_6 alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONRR 9 or C0 2 R 9 and m is 0-2; and particularly, wherein m is 0;
- R 4 is hydrogen, cyclopropyl-Ci- 2 alkyl or Ci_6 alkyl optionally substituted with 1-3 groups selected from cyano, halogen, hydroxy, Ci_6 alkoxy, C0 2 R or NR 2 ; and particularly, wherein R 4 is methyl, ethyl, isopropyl, propyl, or cyanomethyl;
- R 5 and R 6 are independently hydrogen, Ci_6 alkyl or haloCi_6 alkyl; or R 5 and R 6 together with the carbon atoms to which they are attached form a C3-7 cycloalkyl or 5-6 membered heterocyclyl containing 1-2 heteroatoms selected from N, O and S; and particularly, wherein R 5 is hydrogen and R 6 is hydrogen or methyl;
- R 7 is phenyl, 5-6 membered heteroaryl containing 1-3 heteroatoms selected from N, O and S; 5-6 membered heterocyclyl containing 1-2 heteroatoms selected from N, O and S; or a bicyclic 9-10 membered aryl or heteroaryl, said heteroaryl containing 1-3 heteroatoms selected from N, O and S; wherein R 7 is optionally substituted with R 11 and 1-2 R 12 ; and particularly, wherein R 7 is phenyl optionally substituted with R 11 and 1-2 R 12 ; and wherein R 12 is halogen or Ci_6 alkyl;
- R 8 is hydrogen, halogen, cyano, Ci_6alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, C(0)NRR 10 or C0 2 R 10 ; and particularly, wherein R 8 is hydrogen;
- R 9 is Ci-6 alkyl, haloCi_6 alkyl, C3-7 cycloalkyl or phenyl; wherein said C3-7 cycloalkyl, phenyl is optionally substituted with Ci_6 alkyl, C0 2 R or tetrazolyl; and particularly, wherein R 9 is Ci_6 alkyl, haloCi_6 alkyl, cyclopropyl, phenyl optionally substituted with Ci_6 alkyl;
- R 10 is Ci_6 alkyl or haloCi_ 6 alkyl
- R 11 is halo gen or Ci_6 alkyl; and particularly, wherein R 11 is chloro or methyl; m) R is halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkyl, haloCi_6 alkoxy or cyano; and particularly, wherein R 12 is fluoro;
- n) R is H or Ci_6 alkyl
- R a is H, Ci_6 alkyl, hydroxy, halo or Ci_3 alkoxy; and particularly, wherein R a is H or d_ 6 alkyl.
- the invention provides a compound of Formula (8), (9A), (9B), (9C), (9D), (10A) or (10B), wherein R 2 is hydrogen, methoxy or fluoro; and m is 0.
- R 5 is hydrogen;
- R 6 is hydrogen or methyl;
- R 8 is hydrogen.
- R 12 is fluoro.
- the term "compound(s) of the present invention” refers to a compound of Formula (1), (2), (3), (4), (5), (5 A), (5'), (5B), (5C), (6), (7), (8), (9 A), (9B), (9C), (9D), (10A), (10B) or any combination thereof, prodrugs thereof, salts of the compound and/or prodrugs, hydrates or solvates of the compounds, salts and/or prodrugs, as well as all stereoisomers (including diastereoisomers and enantiomers), tautomers and isotopically labeled compounds (including deuterium substitutions), as well as inherently formed moieties (e.g., polymorphs, solvates and/or hydrates).
- Certain of the compounds described herein contain one or more asymmetric centers or axes and may thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)-.
- the present invention is meant to include all possible isomers, including racemic mixtures, optically pure forms and intermediate mixtures.
- Optically active (R)- and (S)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques. If the compound contains a double bond, the substituent may be E or Z configuration. If the compound contains a disubstituted cycloalkyl, the cycloalkyl substituent may have a cis- or trans-configuration. All tautomeric forms are also intended to be included.
- any formula given herein is also intended to represent unlabeled forms as well as isotopically labeled forms of the compounds.
- Isotopically labeled compounds have structures depicted by the formulas given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as 2 H, 3 H, n C, 13 C, 14 C, 15 N, 18 F, 31 P, 32 P, 35 S, 36 C1 and
- the invention includes various isotopically labeled compounds as defined herein, for example those into which radioactive isotopes, such as 3 H, 13 C, and 14 C , are present.
- isotopically labelled compounds are useful in metabolic studies (with 14 C), reaction kinetic studies (with, for example 2 H or 3 H), detection or imaging techniques, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT) including drug or
- Isotopically labeled compounds of this invention and prodrugs thereof can generally be prepared by carrying out the procedures disclosed in the schemes or in the examples and preparations described below by substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent.
- substitution with heavier isotopes may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements or an improvement in therapeutic index.
- deuterium in this context is regarded as a substituent of a compound of the formula (I).
- concentration of such a heavier isotope, specifically deuterium may be defined by the isotopic enrichment factor.
- isotopic enrichment factor as used herein means the ratio between the isotopic abundance and the natural abundance of a specified isotope.
- a substituent in a compound of this invention is denoted deuterium, such compound has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium incorporation at each designated deuterium atom), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium incorporation), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5% deuterium incorporation).
- Isotopically-labeled compounds of the invention can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Processes using an appropriate isotopically- labeled reagents in place of the non-labeled reagent previously employed.
- compositions in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D 2 0, d 6 -acetone, d 6 - DMSO.
- Compounds of the invention that contain groups capable of acting as donors and/or acceptors for hydrogen bonds may be capable of forming co-crystals with suitable co-crystal formers. These co-crystals may be prepared from the compounds of the invention by known co- crystal forming procedures. Such procedures include grinding, heating, co-subliming, co- melting, or contacting in solution the compounds of the invention with the co-crystal former under crystallization conditions and isolating co-crystals thereby formed. Suitable co-crystal formers include those described in WO 2004/078163. Hence the invention further provides co- crystals comprising a compound of the invention.
- any asymmetric atom (e.g., carbon or the like) of the compound(s) of the present invention can be present in racemic or enantiomerically enriched, for example the (R)-, (S)- or (R,S)- configuration.
- each asymmetric atom has at least 50 % enantiomeric excess, at least 60 % enantiomeric excess, at least 70 % enantiomeric excess, at least 80 % enantiomeric excess, at least 90 % enantiomeric excess, at least 95 % enantiomeric excess, or at least 99 % enantiomeric excess in the (R)- or (S)- configuration.
- Substituents at atoms with unsaturated bonds may, if possible, be present in cis- (Z)- or trans- (E)- form.
- a compound of the present invention can be in the form of one of the possible isomers, rotamers, atropisomers, tautomers or mixtures thereof, for example, as substantially pure geometric (cis or trans) isomers, diastereomers, optical isomers (antipodes), racemates or mixtures thereof.
- Any resulting mixtures of isomers can be separated on the basis of the physicochemical differences of the constituents, into the pure or substantially pure geometric or optical isomers, diastereomers, racemates, for example, by chromatography and/or fractional crystallization.
- Any resulting racemates of final products or intermediates can be resolved into the optical antipodes by known methods, e.g., by separation of the
- a basic moiety may thus be employed to resolve the compounds of the present invention into their optical antipodes, e.g., by fractional crystallization of a salt formed with an optically active acid, e.g., tartaric acid, dibenzoyl tartaric acid, diacetyl tartaric acid, di-0,0'-p-toluoyl tartaric acid, mandelic acid, malic acid or camphor- 10-sulfonic acid. Racemic products can also be resolved by chiral chromatography, e.g., high pressure liquid chromatography (HPLC) using a chiral adsorbent.
- HPLC high pressure liquid chromatography
- the compounds of the invention in free form or in salt form exhibit valuable pharmacological properties, e.g. LPA modulating properties, e.g. as indicated in in vitro and/or in vivo tests as provided in the next sections, and are therefore indicated for therapy in treating a disorder which may be treated by modulating LPA, such as those described below.
- LPA modulating properties e.g. as indicated in in vitro and/or in vivo tests as provided in the next sections, and are therefore indicated for therapy in treating a disorder which may be treated by modulating LPA, such as those described below.
- the compounds of Formula (1), (2), (3), (4), (5), (5 A), (5'), (5B), (5C), (6), (7), (8), (9 A), (9B), (9C), (9D), (10A) or (10B), are antagonists of at least one of the LPA receptors selected from LPAl, LPA2, LPA3, LPA4, LPA5 and LPA6, and more particularly antagonists of LPAl.
- the compounds of the invention or a pharmaceutically acceptable salt thereof are useful in the treatment of diseases, disorders, or conditions in which activation of at least one LPA receptor by LPA contributes to the symptomology or progression of the disease, disorder or condition.
- diseases, disorders, or conditions may arise from one or more of a genetic, iatrogenic, immunological, infectious, metabolic, oncological, toxic, surgical, and/or traumatic etiology.
- medicaments described herein comprise antagonists of LPA receptors.
- the methods, compounds, pharmaceutical compositions, and medicaments described herein comprise antagonists of LPAl.
- the compounds of the invention are useful for the treatment of a LPA-dependent or LPA-mediated disease or condition selected from pulmonary fibrosis, idiopathic pulmonary fibrosis, a diffuse parenchymal interstitial lung disease including iatrogenic drug-induced fibrosis, occupational and/or environmental induced fibrosis (Farmer lung), granulomatous diseases (sarcoidosis, hypersensitivity pneumonia), collagen vascular disease (scleroderma), alveolar proteinosis, langerhans cell granulomatosis,
- pulmonary fibrosis idiopathic pulmonary fibrosis
- a diffuse parenchymal interstitial lung disease including iatrogenic drug-induced fibrosis, occupational and/or environmental induced fibrosis (Farmer lung), granulomatous diseases (sarcoidosis, hypersensitivity pneumonia), collagen vascular disease (scleroderma), alveolar proteinosis, langerhans cell granulomatosis,
- lymphangioleiomyomatosis Hermansky-Pudlak Syndrome, tuberous sclerosis
- the compounds of the invention or a pharmaceutically acceptable salt thereof are useful for the treatment of a LPA-dependent or LPA-mediated disease or condition selected from fibrosis of organs or tissues, scarring, liver diseases, dermatological conditions, cancer, cardiovascular disease, respiratory diseases or conditions including asthma, chronic obstructive pulmonary disease (COPD), pulmonary fibrosis, pulmonary arterial hypertension or acute respiratory distress syndrome; inflammatory disease, gastrointestinal tract disease, renal disease, urinary tract-associated disease, inflammatory disease of lower urinary tract, dysuria, frequent urination, pancreas disease, arterial obstruction, cerebral infarction, cerebral hemorrhage, pain, peripheral neuropathy, and fibromyalgia.
- a LPA-dependent or LPA-mediated disease or condition selected from fibrosis of organs or tissues, scarring, liver diseases, dermatological conditions, cancer, cardiovascular disease, respiratory diseases or conditions including asthma, chronic obstructive pulmonary disease (COPD), pulmonary fibrosis, pulmonary arterial hypertension or acute respiratory distress syndrome;
- the compounds of the invention are useful for the treatment of a LPA-dependent or LPA-mediated disease or condition selected from radiation induced fibrosis; chronic obstructive pulmonary disease (COPD), scleroderma, systemic sclerosis, bleomycin induced pulmonary fibrosis, chronic asthma, silicosis, asbestos induced pulmonary fibrosis, acute respiratory distress syndrome (ARDS), kidney fibrosis, tubulointerstitium fibrosis, glomerular nephritis, focal segmental glomerular sclerosis, lupus nephritis, IgA nephropathy, hypertension, Alport, gut fibrosis, liver fibrosis, cirrhosis, alcohol induced liver fibrosis, toxic/drug induced liver fibrosis, hemochromatosis, nonalcoholic steatohepatitis (NASH), biliary duct injury; primary biliary cirrhosis, infection
- COPD chronic ob
- the compounds of the invention or a pharmaceutically acceptable salt thereof are useful for the treatment of a LPA-dependent or LPA-mediated disease or condition selected from fibrosis of organs (liver, kidney, lung, heart and the like), liver diseases (acute hepatitis, chronic hepatitis, liver fibrosis, liver cirrhosis, portal hypertension, regenerative failure, liver hypofunction, hepatic blood flow disorder, and the like), cell proliferative disease (cancer (solid tumor, solid tumor metastasis, vascular fibroma, myeloma, multiple myeloma, Kaposi's sarcoma, leukemia, chronic lymphocytic leukemia (CLL and the like) and invasive metastasis of cancer cell, and the like), inflammatory disease (psoriasis, nephropathy, pneumonia and the like), gastrointestinal tract disease (irritable bowel syndrome (IBS), inflammatory bowel disease (IBD), abnormal pancreatic
- the compounds of the invention or a pharmaceutically acceptable salt thereof are useful for the treatment of a LPA-dependent or LPA-mediated disease or condition selected from renal fibrosis (tubulointerstitium fibrosis, glomerular sclerosis), acute kidney injury, chronic kidney disease, skin fibrosis, fibrosis of the gut, ocular fibrosis, breast cancer, pancreatic cancer, ovarian cancer, prostate cancer, glioblastoma, bone cancer, colon cancer, bowel cancer, head and neck cancer, melanoma, multiple myeloma, chronic lymphocytic leukemia, cancer pain, tumor metastatis, transplant organ rejection, age related macular degeneration (AMD), diabetic retinopathy, and Raynaud's phenomenon.
- a LPA-dependent or LPA-mediated disease or condition selected from renal fibrosis (tubulointerstitium fibrosis, glomerular sclerosis), acute kidney injury, chronic kidney disease, skin fibrosis, fibrosis of the
- Compounds disclosed herein are also useful for the treatment of post-transplant fibrosis associated with chronic rejection (bronchiolitis obliterans for lung transplant), cutaneous fibrosis (cutaneous scleroderma, Dupuytren disease, keloids), hepatic fibrosis with or without cirrhosis (toxic/drug induced hemochromatosis), alcoholic liver disease, viral hepatitis (hepatitis B virus, hepatitis C virus, HCV), metabolic and auto-immune diseases.
- the invention provides a method for the treatment or prevention of pulmonary fibrosis (idiopathic pulmonary fibrosis), renal fibrosis, hepatic fibrosis, ocular fibrosis, or cardiac fibrosis in a cell, tissue or subject in need thereof, comprising administering a therapeutically effective amount of a compound of Formula (1), (2), (3), (4), (5), (5 A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B), or a pharmaceutically acceptable salt thereof to said subject.
- Pulmonary Fibrosis idiopathic pulmonary fibrosis
- renal fibrosis hepatic fibrosis
- ocular fibrosis hepatic fibrosis
- cardiac fibrosis in a cell, tissue or subject in need thereof
- Pulmonary Fibrosis idiopathic pulmonary fibrosis
- renal fibrosis hepatic fibro
- LPA is an important mediator of fibroblast recruitment in pulmonary fibrosis.
- LPA and LPA1 play key pathogenic roles in pulmonary fibrosis.
- Fibroblast chemo-attractant activity plays an important role in the lungs in patients with pulmonary fibrosis.
- Profibrotic effects of LPA1 -receptor stimulation is explained by LPA1 -receptor-mediated vascular leakage and increased fibroblast recruitment, both profibrotic events.
- the LPA-LPA1 pathway has a role in mediating fibroblast migration and vascular leakage in IPF. The end result is the aberrant healing process that characterizes this fibrotic condition.
- the LPA1 receptor is the LPA receptor most highly expressed on fibroblasts obtained from patients with IPF. Furthermore, bronchoalveolar lavage (BAL) obtained from IPF patients induced chemotaxis of human fetal lung fibroblasts that was blocked by the dual LPA1- LP A3 receptor antagonist Ki 16425. In an experimental bleomycin- induced lung injury mouse model, it was shown that LPA levels were high in BAL samples compared with unexposed controls. LPA1 knockout mice are protected from fibrosis after bleomycin challenge with reduced fibroblast accumulation and vascular leakage. In human subjects with IPF, high LPA levels were observed in bronchoalveolar lavage samples compared with healthy controls.
- LPA and LPA1 are involved in the etiology of kidney fibrosis. LPA has effects on both proliferation and contraction of glomerular mesangial cells and thus has been implicated in proliferative glomerulonephritis (Inoue et al., Clinical Science 96, 431- 436, 1999).
- LPA LPA1 receptor
- LPA can participate in intraperitonial accumulation of monocyte/macrophages and that LPA can induce expression of the profibrotic cytokine CTGF in primary cultures of human fibroblasts (Koh et al., J. Clin. Invest. 102 (1998) 716-727).
- LPA treatment of a mouse epithelial renal cell line, MCT induced a rapid increase in the expression of the profibrotic cytokine CTGF.
- CTGF plays a crucial role in UUO- induced tubulointerstitial fibrosis (TIF), and is involved in the profibrotic activity of TGF beta. This induction was almost completely suppressed by co-treatment with the LPA-receptor antagonist Kil6425.
- the profibrotic activity of LPA in kidney results from a direct action of LPA on kidney cells involving induction of CTGF.
- LPA is implicated in liver disease and fibrosis. Plasma LPA levels and serum autotoxin (enzyme responsible for LPA production) are elevated in hepatitis patients and animal models of liver injury in correlation with increased fibrosis. LPA also regulates liver cell function. LPA1 and LPA2 receptors are expressed by mouse hepatic stellate cells and LPA stimulates migration of hepatic myofibroblasts.
- LPA is in involved in wound healing in the eye.
- LPA1 and LPA 3 receptors are detectable in the normal rabbit corneal epithelial cells, keratocytes and endothelial cells and LPA1 and LPA 3 expression are increased in corneal epithelial cells following injury.
- LPA and its homologues are present in the aqueous humor and the lacrimal gland fluid of the rabbit eye and these levels are increased in a rabbit corneal injury model.
- LPA induces actin stress fiber formation in rabbit corneal endothelial and epithelial cells and promotes contraction corneal fibroblasts.
- LPA also stimulates proliferation of human retinal pigmented epithelial cells Cardiac fibrosis
- LPA is implicated in myocardial infarction and cardiac fibrosis. Serum LPA levels are increased in patients following myocardial infarction (MI) and LPA stimulates proliferation and collagen production (fibrosis) by rat cardiac fibroblasts. Both LPAland LPA3 receptors are highly expressed in human heart tissue.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the present invention or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the pharmaceutical composition can be formulated for particular routes of administration such as oral administration, parenteral administration, and rectal administration, etc.
- the pharmaceutical compositions of the present invention can be made up in a solid form (including without limitation capsules, tablets, pills, granules, powders or suppositories), or in a liquid form (including without limitation solutions, suspensions or emulsions).
- compositions can be subjected to conventional pharmaceutical operations such as sterilization and/or can contain conventional inert diluents, lubricating agents, or buffering agents, as well as adjuvants, such as preservatives, stabilizers, wetting agents, emulsifers and buffers, etc.
- the pharmaceutical compositions are tablets or gelatin capsules comprising the active ingredient together with
- diluents e.g., lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycine
- lubricants e.g., silica, talcum, stearic acid, its magnesium or calcium salt and/or poly ethylenegly col; for tablets also
- diluents e.g., lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycine
- lubricants e.g., silica, talcum, stearic acid, its magnesium or calcium salt and/or poly ethylenegly col
- binders e.g. , magnesium aluminum silicate, starch paste, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone; if desired
- disintegrants e.g. , starches, agar, alginic acid or its sodium salt, or effervescent mixtures; and/or
- Tablets may be either film coated or enteric coated according to methods known in the art.
- compositions for oral administration include an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof, in the form of tablets, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft capsules, or syrups or elixirs.
- Compositions intended for oral use are prepared according to any method known in the art for the manufacture of pharmaceutical compositions and such compositions can contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide
- Tablets may contain the active ingredient in admixture with nontoxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients are, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example, starch, gelatin or acacia; and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets are uncoated or coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monostearate or glyceryl distearate can be employed.
- Formulations for oral use can be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example, peanut oil, liquid paraffin or olive oil.
- compositions are aqueous isotonic solutions or suspensions, and suppositories are advantageously prepared from fatty emulsions or suspensions.
- Said compositions may be sterilized and/or contain adjuvants, such as preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers. In addition, they may also contain other therapeutically valuable substances.
- Said compositions are prepared according to conventional mixing, granulating or coating methods, respectively, and contain about 0.1-75%, or contain about 1-50%, of the active ingredient.
- Suitable compositions for transdermal application include an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof, with a suitable carrier.
- Carriers suitable for transdermal delivery include absorbable pharmacologically acceptable solvents to assist passage through the skin of the host.
- transdermal devices are in the form of a bandage comprising a backing member, a reservoir containing the compound optionally with carriers, optionally a rate controlling barrier to deliver the compound of the skin of the host at a controlled and predetermined rate over a prolonged period of time, and means to secure the device to the skin.
- Suitable compositions for topical application e.g.
- aqueous solutions, suspensions, ointments, creams, gels or sprayable formulations e.g. , for delivery by aerosol or the like.
- topical delivery systems will in particular be appropriate for dermal application, e.g. , for the treatment of skin cancer, e.g. , for prophylactic use in sun creams, lotions, sprays and the like. They are thus particularly suited for use in topical, including cosmetic, formulations well-known in the art.
- Such may contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
- a topical application may also pertain to an inhalation or to an intranasal application. They may be conveniently delivered in the form of a dry powder (either alone, as a mixture, for example a dry blend with lactose, or a mixed component particle, for example with phospholipids) from a dry powder inhaler or an aerosol spray presentation from a pressurised container, pump, spray, atomizer or nebuliser, with or without the use of a suitable propellant.
- a dry powder either alone, as a mixture, for example a dry blend with lactose, or a mixed component particle, for example with phospholipids
- Dosage forms for the topical or transdermal administration of a compound of this invention include powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches and inhalants.
- the active compound may be mixed under sterile conditions with a
- pharmaceutically acceptable carrier and with any preservatives, buffers, or propellants that may be desirable.
- the ointments, pastes, creams and gels may contain, in addition to an active compound of this invention, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- excipients such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- Powders and sprays can contain, in addition to a compound of this invention, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances.
- Sprays can additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
- Transdermal patches have the added advantage of providing controlled delivery of a compound of the present invention to the body.
- dosage forms can be made by dissolving or dispersing the compound in the proper medium.
- Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate of such flux can be controlled by either providing a rate controlling membrane or dispersing the active compound in a polymer matrix or gel.
- Ophthalmic formulations are also contemplated as being within the scope of this invention.
- the present invention further provides anhydrous pharmaceutical compositions and dosage forms comprising the compounds of the present invention or a pharmaceutically acceptable salt thereof as active ingredients, since water may facilitate the degradation of certain compounds.
- Anhydrous pharmaceutical compositions and dosage forms of the invention can be prepared using anhydrous or low moisture containing ingredients and low moisture or low humidity conditions.
- An anhydrous pharmaceutical composition may be prepared and stored such that its anhydrous nature is maintained. Accordingly, anhydrous compositions are packaged using materials known to prevent exposure to water such that they can be included in suitable formulary kits. Examples of suitable packaging include, but are not limited to, hermetically sealed foils, plastics, unit dose containers (e. g. , vials), blister packs, and strip packs.
- compositions and dosage forms that comprise one or more agents that reduce the rate by which the compound of the present invention as an active ingredient will decompose.
- agents which are referred to herein as “stabilizers,” include, but are not limited to, antioxidants such as ascorbic acid, pH buffers, or salt buffers, etc.
- the pharmaceutical composition or combination of the present invention can be in unit dosage of about 1-1000 mg of active ingredient(s) for a subject of about 50-70 kg, or about 1-500 mg or about 1-250 mg or about 1-150 mg or about 0.5-100 mg, or about 1-50 mg of active ingredients.
- the therapeutically effective dosage of a compound, the pharmaceutical composition, or the combinations thereof is dependent on the species of the subject, the body weight, age and individual condition, the disorder or disease or the severity thereof being treated. A physician, clinician or veterinarian of ordinary skill can readily determine the effective amount of each of the active ingredients necessary to prevent, treat or inhibit the progress of the disorder or disease.
- the above-cited dosage properties are demonstrable in vitro and in vivo tests using advantageously mammals, e.g. , mice, rats, dogs, monkeys or isolated organs, tissues and preparations thereof.
- the compounds of the present invention or a pharmaceutically acceptable salt thereof can be applied in vitro in the form of solutions, e.g. , aqueous solutions, and in vivo either enterally, parenterally, advantageously intravenously, e.g. , as a suspension or in aqueous solution.
- the dosage in vitro may range between about 10 ⁇ 3 molar and 10 ⁇ 9 molar
- a therapeutically effective amount in vivo may range depending on the route of administration, between about 0.1-500 mg/kg, or between about 1-100 mg/kg.
- the compound of the present invention or a pharmaceutically acceptable salt thereof may be administered either simultaneously with, or before or after, one or more other therapeutic agent.
- the compound of the present invention may be administered separately, by the same or different route of administration, or together in the same pharmaceutical composition as the other agents.
- the invention provides a product comprising a compound of Formula (1), (2), (3), (4), (5), (5A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B) or a pharmaceutically acceptable salt thereof, and at least one other therapeutic agent as a combined preparation for simultaneous, separate or sequential use in the treatment of a disease or condition mediated by LPA.
- Products provided as a combined preparation include: a composition comprising a compound of the invention or a pharmaceutically acceptable salt thereof, and the other therapeutic agent(s) together in the same pharmaceutical composition; or a compound of the invention or a pharmaceutically acceptable salt thereof, and the other therapeutic agent(s) in separate form, e.g. in the form of a kit.
- the pharmaceutical composition may comprise a pharmaceutically acceptable excipient, as described above.
- the invention provides a combination of a compound of the invention or a pharmaceutically acceptable salt thereof, and one or more additional
- therapeutically active agents selected from: corticosteroids, immunosuppresants, analgesics, anti-cancer agent, anti-inflammatories, chemokine receptor antagonists, bronchodilators, leukotriene receptor antagonists, leukotriene formation inhibitors, monoacylglycerol kinase inhibitors, phospholipase Al inhibitors, phospholipase A2 inhibitors, and lysophospholipase D (lysoPLD) inhibitors, autotaxin inhibitors, decongestants, antihistamines, mucolytics, anticholinergics, antitussives, expectorants, and ⁇ -2 agonists.
- corticosteroids selected from: corticosteroids, immunosuppresants, analgesics, anti-cancer agent, anti-inflammatories, chemokine receptor antagonists, bronchodilators, leukotriene receptor antagonists, leukotriene formation inhibitors, monoacylgly
- the invention provides a kit comprising two or more separate pharmaceutical compositions, at least one of which contains a compound of the invention or a pharmaceutically acceptable salt thereof.
- the kit comprises means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet.
- An example of such a kit is a blister pack, as typically used for the packaging of tablets, capsules and the like.
- the kit of the invention may be used for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another.
- the kit of the invention typically comprises directions for administration.
- the compound of the invention or a pharmaceutically acceptable salt thereof and the other therapeutic agent may be manufactured and/or formulated by the same or different manufacturers. Moreover, the compound of the invention and the other therapeutic may be brought together into a combination therapy: (i) prior to release of the combination product to physicians (e.g. in the case of a kit comprising the compound of the invention and the other therapeutic agent); (ii) by the physician themselves (or under the guidance of the physician) shortly before administration; (iii) in the patient themselves, e.g. during sequential administration of the compound of the invention and the other therapeutic agent.
- the invention provides the use of a compound of the invention (e.g., Formula (1), (2), (3), (4), (5), (5A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B)) or a pharmaceutically acceptable salt thereof, for treating a disease or condition mediated by LPA, wherein the medicament is prepared for administration with another therapeutic agent.
- a compound of the invention e.g., Formula (1), (2), (3), (4), (5), (5A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B)
- a pharmaceutically acceptable salt thereof for treating a disease or condition mediated by LPA, wherein the medicament is prepared for administration with another therapeutic agent.
- the invention also provides the use of another therapeutic agent for treating a disease or condition mediated by LPA, wherein the medicament is administered with a compound of the invention or a pharmaceutically acceptable salt
- the invention also provides a compound of the invention for use in a method of treating a disease or condition mediated by LPA, wherein a compound of the invention (e.g., Formula (1), (2), (3), (4), (5), (5A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B)) or a pharmaceutically acceptable salt thereof, is prepared for administration with another therapeutic agent.
- a compound of the invention e.g., Formula (1), (2), (3), (4), (5), (5A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B)
- a pharmaceutically acceptable salt thereof is prepared for administration with another therapeutic agent.
- the invention also provides another therapeutic agent for use in a method of treating a disease or condition mediated by LPA, wherein the other therapeutic agent is prepared for administration with a compound of the invention.
- the invention also provides a compound of the invention, for use in a method of treating a disease or condition mediated by LPA, wherein a compound of the invention or a pharmaceutically acceptable salt thereof is administered with another therapeutic agent.
- the invention also provides another therapeutic agent for use in a method of treating a disease or condition mediated by LPA, wherein the other therapeutic agent is administered with a compound of the invention.
- the invention also provides the use of a compound of the invention (e.g., Formula (1), (2), (3), (4), (5), (5A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B)) or a pharmaceutically acceptable salt thereof, for treating a disease or condition mediated by LPA, wherein the patient has previously (e.g. within 24 hours) been treated with another therapeutic agent.
- the invention also provides the use of another therapeutic agent for treating a disease or condition mediated by LPA, wherein the patient has previously (e.g. within 24 hours) been treated with a compound of the invention.
- the compounds of the invention can be prepared according to any one of Schemes 1, 2, 3 or 4, provided infra.
- Each reaction step can be carried out in a manner known to those skilled in the art.
- a reaction can be carried out in the presence of a suitable solvent or diluent or of mixture thereof.
- a reaction can also be carried out, if needed, in the presence of an acid or a base, with cooling or heating, for example in a temperature range from approximately -30 °C to approximately 150 °C.
- a reaction is carried out in a temperature range from approximately 0 °C to 100 °C, and more particularly, in a temperature range from room temperature to approximately 80 °C, in an open or closed reaction vessel and/or in the atmosphere of an inert gas, for example nitrogen.
- an inert gas for example nitrogen.
- Z 1 , Z 2 , R 1 , R 2 , R 3 , R 4a , R 4b , R 5 , R 6 , R 6a ,R 6b , B, m, n and p are as previously defined
- X, X , and X are leaving groups; and R is hydrogen or Ci ⁇ alkyl; or two R groups together with boron can form a cyclic boronate ester.
- a compound of Formula (1) can be prepared by Suzuki reaction of a compound of Formula 1-4 with a compound of Formula 1-5, as described in detail below in Scheme 2a.
- a compound of Formula 1-4 can, in turn, be prepared by reaction of a compound of Formula 1-1 with an alkoxycarbonylation reagent of Formula 1-3.
- Suitable alkoxycarbonylation reagents include chloroformates and dialkyl dicarbonates (for example, di-tert-butyl dicarbonate).
- the reaction generally takes place in the presence of a base such as triethylamine or pyridine, in an inert solvent such as dichloromethane or dioxane, at a temperature of about -78 °C to about rt.
- the reaction can take place in a biphasic system consisting of an inert solvent such as dioxane together with an aqueous basic solution such as aqueous sodium carbonate or sodium bicarbonate.
- a compound of Formula 1-4 can also be prepared by reaction of a compound of Formula 1-1 with phosgene, followed by treatment with an alcohol of Formula 1-2.
- a compound of Formula (1) can also be prepared by reversal of the order of the above reaction steps; that is, Suzuki reaction of a compound of Formula 1-1 with a compound of Formula 1-5, followed by reaction of the resulting compound of Formula 1-6 with phosgene and an alcohol of Formula 1-2.
- An alternative preparation of an intermediate compound of Formula 1-6 can be performed by Suzuki reaction of a compound of Formula 1-8 with a boronic acid derivative of Formula 1-9.
- a compound of Formula 1-8 can in turn be prepared by selective Suzuki reaction of a compound of Formula 1-1 with a halophenylboronic acid derivative (preferably a chlorophenylboronic acid derivative in which X 1 is CI) of Formula 1-7.
- a compound of Formula (1) can also be prepared by Suzuki reaction of a compound of Formulal-9 with a compound of Formula 1-11.
- a compound of Formula 1-11 can in turn be prepared by selective Suzuki reaction of a compound of Formula 1-4 with a halophenylboronic acid derivative (preferably a chlorophenylboronic acid derivative in which X 1 is CI) of Formula 1-7.
- a compound of Formula 1 can be prepared by a Suzuki reaction in which the roles of the coupling partners are reversed; that is, by reaction of a boronic acid derivative of Formula 1-12 with a compound of Formula 1-13.
- a compound of Formula 1-12 can be prepared by treatment of a compound of Formula 1-11 with a diboron compound (for example, bis- pinacolatodiboron) under palladium catalysis.
- a compound of Formula (1) can also be prepared by reaction of a compound of Formula 1-16 with DPP A, in the presence of a compound of Formula 1-2 and a base.
- a compound of Formula 1-16 can in turn be prepared by Suzuki coupling of a compound of Formula 1-15 with a compound of Formula 1-9, followed by ester hydrolysis.
- a compound of Formula 1-15 can be prepared by cycloaddition reaction of a compound of Formula 1-14 with a diazoacetate ester, followed by alkylation with an alkylating agent R 5 X, in the presence of a base such as sodium hydride.
- a compound of Formula I- 11 can also be prepared by ester hydrolysis of a compound of Formula 1-15, followed by reaction with DPPA, in the presence of a compound of Formula 1-2 and a base.
- Compounds of Formula 1-1, 1-2, 1-3, 1-5, 1-7, 1-9, 1-13, and 1-14 are commercially available, or can be prepared by literature methods or by methods available to those skilled in the art.
- compounds of Formula (2) can be prepared following the
- compounds of Formula (2) can be prepared by coupling a compound of Formula 2a-2, in which X is a leaving group (e.g., iodo, bromo, chloro, trifluoromethane- sulfonyloxy, and the like), with a compound of Formula 1-5.
- X is a leaving group (e.g., iodo, bromo, chloro, trifluoromethane- sulfonyloxy, and the like), with a compound of Formula 1-5.
- the reaction takes place in the presence of a suitable transition metal catalyst (e.g., tetrakis(triphenylphosphinepalladium)(0), PdCl 2 (dppf)), or dichloro [ ⁇ , ⁇ bis(di-tert- butylphosphino)]ferrocene palladium (II)), a suitable solvent (e.g., DME, dioxane, toluene, ethanol, and the like) and a suitable base (e.g., anhydrous potassium carbonate or aqueous sodium carbonate solution, and the like).
- a suitable transition metal catalyst e.g., tetrakis(triphenylphosphinepalladium)(0), PdCl 2 (dppf)
- a suitable solvent e.g., DME, dioxane,
- reaction mixture is optionally further reacted to remove any protecting groups.
- organometallic coupling reactions for example using tin reagents (Stille coupling) or zinc reagents (Negishi coupling), may also be employed in place of the Suzuki coupling reaction using boron reagents described in Scheme 2a.
- Intermediate compounds of Formula 2a-2 can be prepared by a reaction of a compound of Formula 2-1 with phosgene, followed by reaction with an alcohol of Formula 2a- 1.
- the reaction takes place in the presence of a suitable base such as pyridine, optionally in the presence of a drying agent such as molecular sieves, in an inert solvent such as dichloromethane, THF, and the like, at a temperature from about 20 °C to about 65 °C.
- compounds of Formula (2) can be prepared by an alternate sequence in which the order of reaction steps is reversed.
- Suzuki reaction of a compound of Formula 2-1 with a compound of Formula 1-5 provides an intermediate compound of Formula 1-6, which can then be reacted with DPPA and a compound of Formula 2a- 1 as described above to furnish a compound of Formula (2).
- Intermediate compounds of Formula 1-6 can also be prepared by Suzuki reaction of a compound of Formula 1-9 with a compound of Formula 2-8.
- Compounds of Formula (2) can be prepared by Suzuki coupling of a compound of Formula 2b- 1 with a compound of Formula 1-9, using similar methods to those described in Scheme 2a.
- Compounds of Formula (2) can also be prepared by Suzuki reaction in which the roles of the coupling partners are reversed; that is, by reaction of a boronic acid derivative of Formula 2b-2 with a compound of Formula 1-13.
- a compound of Formula 2b-2 can be prepared by treatment of a compound of Formula 2b- 1 with a diboron compound (for example, bis(pinacolato)diboron and the like) in the presence of a suitable transition metal catalyst (for example PdCl 2 (dppf)),) and a suitable base (for example, potassium acetate and the like) in a suitable solvent (for example, toluene, dioxane and the like).
- a suitable transition metal catalyst for example PdCl 2 (dppf)
- a suitable base for example, potassium acetate and the like
- a suitable solvent for example, toluene, dioxane and the like.
- Z 2 , R 1 , R 2 , R 3 , R 4a , R 4b , R 5 , R 6 , R 7 , R 8 , B, m, n and p are as previously defined in Formula (1).
- compounds of Formula (2) or 2b- 1 can be prepared by reaction of a carboxylic acid compound of Formula 4- 1 with diphenylphosphoryl azide (DPP A) and an alcohol of Formula 2a- 1, in an inert solvent such as dichloromethane, in the presence of a base such as triethylamine or DIPEA. If desired, a compound of Formula (2) can be converted to another compound of Formula (2), or a compound of Formula 2b- 1 can be converted to another compound of Formula 2b- 1.
- DPP A diphenylphosphoryl azide
- DIPEA inert solvent
- a base such as triethylamine or DIPEA
- compounds of Formula 2-1 can be prepared by halogenation of a pyrazole compound of Formula 2- lb.
- appropriate halogenating agents include N-bromosuccinimide and Br 2 ;
- X is iodine, appropriate halogenating agents include N-iodosuccinimide;
- appropriate reagents include N-chlorosuccinimide and Cl 2 .
- the reaction takes place in a suitable solvent such as dichloromethane, at a temperature from about -78 °C to about 40 °C.
- a compound of Formula 2- lb can be prepared by reaction of a compound of Formula 2- la with an alkylating agent R 5 X' , in which X' is a halogen such as chloro, bromo, or iodo, or an alkyl- or aryl-sulfonyloxy group.
- the alkylation takes place in the presence of a suitable base such as sodium hydride, potassium carbonate, or the like, in a suitable solvent such as DMF or DMSO, at a temperature of about 0 °C to about 120 °C.
- a suitable base such as sodium hydride, potassium carbonate, or the like
- a suitable solvent such as DMF or DMSO
- An intermediate compound of Formula 6-3 is a particular embodiment of a compound of Formula 2-8 in which X 1 is CI. Such compounds can be prepared as shown in
- a compound Formula 6-3 can be prepared by coupling a compound of Formula 6-1 (i.e., a compound of Formula 1-1 in which X is Br and Z 1 is CH), with a compound of Formula 6-2.
- the reaction takes place in the presence of a suitable transition metal catalyst (e.g., tetrakis(triphenylphosphinepalladium)(0), PdCi 2 (dppf)), or dichloro [ ⁇ , bis(di-tert- butylphosphino)]ferrocene palladium (II)), a suitable solvent (e.g., DME, dioxane, toluene, ethanol, and the like) and a suitable base (e.g., anhydrous potassium carbonate or aqueous sodium carbonate solution, and the like).
- a suitable transition metal catalyst e.g., tetrakis(triphenylphosphinepalladium)(0), PdCi 2 (dppf)
- the reaction proceeds in a temperature range of about 20°C to about 120°C and can take up to about 24 hours to complete.
- the reaction mixture is optionally further reacted to remove any protecting groups.
- organometallic coupling reactions for example using tin reagents (Stille coupling) or zinc reagents (Negishi coupling), may also be employed in place of the boron reagents used in the Suzuki coupling reaction described in Scheme 6.
- X 1 CI Scheme 7 wherein Z 2 , R 1 , R 2 , R 3 , R 4a , R 4b , R 5 , R 6 , R 7 , R 8 , B, m, n and p are as previously defined in Formula (2); X and X 1 are leaving groups; and R 20 is as defined in Scheme 1.
- compounds of Formula 2b- 1 in which X 1 is CI, Br, or I can be prepared by Sandmeyer reaction of an aniline compound of Formula 7-3.
- the reaction is typically performed using sodium nitrite in acidic aqueous medium, or an alkyl nitrite in aqueous or nonaqueous solution, in the presence of a copper(I) or copper(II) halide salt.
- the reaction can be performed using copper(II) bromide and an alkyl nitrite such as tert-butyl nitrite, in a suitable solvent such as anhydrous acetonitrile, at a temperature of about rt, for a time of about 1-4 h.
- a compound of Formula 2b- 1 in which X 1 is triflate can be prepared by reaction of a compound of Formula 7-4 with trifluoromethanesulfonic anhydride, in an inert solvent such as dichloromethane, in the presence of a base such as N,N-dimethylaminopyridine or pyridine, at a temperature of about -78 °C to about rt.
- Compounds of Formula 7-3 and 7-4 can in turn be prepared by Suzuki coupling of a compound of Formula 7-1 or 7-2, respectively, with a compound of Formula 2a-2 as described earlier in Scheme 2a. For these reactions, the amino and hydroxyl groups may optionally be protected with a protecting group P.
- a compound of Formula 2b- 1 in which X 1 is CI can be prepared directly via Suzuki coupling of a compound of Formula 2a-2 with a chlorophenylboronate compound of Formula 6-2.
- E is (R )n ; or E is OH, OP, Br, CI, or I, wherein P is a protecting group; and Z 2 , R 1 , R 2 , R 3 , R 4a , R 4b , R 5 , R 6 , R 7 , R 8 , B, m, n and p are as previously defined in Formula (2).
- a carboxylic acid of Formula 4-1 can be prepared by hydrolysis of an ester compound of Formula 8-3a using acidic or basic methods known to those skilled in the art; for example, by treatment with lithium hydroxide in a mixture of THF and water, at about 0 °C to about 65 °C.
- An ester compound of Formula 8-3a (or an analogous lower alkyl ester) can be prepared via alkylation of a pyrazole compound of Formula 8-2 with an alkylating agent R 5 X', in which X' is a halogen such as chloro, bromo, or iodo, or an alkyl- or aryl-sulfonyloxy group.
- the alkylation takes place in the presence of a suitable base such as sodium hydride, potassium carbonate, or the like, in a suitable solvent such as DMF or DMSO, at a temperature of about 0 °C to about 120 °C.
- a suitable base such as sodium hydride, potassium carbonate, or the like
- a suitable solvent such as DMF or DMSO
- the isomeric product of Formula 8-3b may be formed.
- Compounds of Formulas 8-3a and 8-3b can be separated by those skilled in the art using techniques such as silica gel chromatography, crystallization, or reverse-phase preparative HPLC.
- a compound of Formula 8-2 can be prepared by reaction of a nitroalkene compound of Formula 8-1 with a diazoacetate ester such as ethyl diazoacetate. The reaction takes place in an inert solvent such as THF or toluene, at a temperature of about 20 °C to about 150 °C.
- intermediate compounds of Formula 8-2 can be prepared by reaction of an alkyne compound of Formula 8-4 with a diazoacetate ester such as ethyl diazoacetate. The reaction takes place in an inert solvent such as THF or toluene, at a temperature of about 20 °C to about 150 °C. During this reaction the isomeric product of Formula 8-5 may be formed.
- Compounds of Formulas 8-2 and 8-5 can be separated by those skilled in the art using techniques such as silica gel chromatography, crystallization, or reverse-phase preparative HPLC.
- R , R , R , and m are as previously defined in Formula (1); and X is CI, Br, I.
- a compound of Formula 9-3 can be prepared by reaction of a compound of Formula 9-2 with a diboron compound (for example, bis(pinacolato)diboron and the like) in the presence of a suitable transition metal catalyst (for example PdCl 2 (dppf)),) and a suitable base (for example, potassium acetate and the like) in a suitable solvent (for example, toluene, dioxane and the like).
- a suitable transition metal catalyst for example PdCl 2 (dppf)
- a suitable base for example, potassium acetate and the like
- a suitable solvent for example, toluene, dioxane and the like
- a compound of Formula 9-2 can in turn be prepared by alkylation of a compound of Formula 9-1 with a bis-electrophile (for example, 1,2- dibromoe thane).
- the reaction takes place in the presence of a suitable base (for example, sodium hydride) in a solvent such as DMF, at a temperature of about 0 °C to about 100 °C.
- a suitable base for example, sodium hydride
- the reaction can take place under phase transfer conditions using an organic solvent such as toluene or dichloromethane, an aqueous base such as sodium or potassium hydroxide solution, and a phase transfer agent such as tetrabutylammonium bromide, at a temperature of about 0 °C to about 50 °C.
- a suitable base for example, sodium hydride
- a solvent such as DMF
- the reaction can take place under phase transfer conditions using an organic solvent such as toluene or dichloromethane, an aqueous base such as sodium
- R 1 , R 2 , R 3 , R 4a , R 4b , R 5 , R 6 , R 6a , R 6b , B, m, n and p are as previously defined in
- a compound of Formula 10-10 can be prepared by a metal-catalyzed cross-coupling reaction of a compound of Formula 10-9 with a boronic acid such as a substituted phenyl boronic acid of Formula 1-9, in the presence of a palladium catalyst such as dichloro [ ⁇ , ⁇ bis(di-tert-butylphosphino)]ferrocene palladium (II), a base such as potassium phosphate tribasic in an inert solvent such as 1 ,4-dioxane with heating at elevated temperatures either thermally or with microwave irradiation.
- a palladium catalyst such as dichloro [ ⁇ , ⁇ bis(di-tert-butylphosphino)]ferrocene palladium (II)
- a base such as potassium phosphate tribasic in an inert solvent such as 1 ,4-dioxane with heating at elevated temperatures either thermally or with microwave irradiation.
- Intermediate compounds of Formula 10-9 can be prepared by Curtis rearrangement of a carboxylic acid of Formula 10-7 in the presence of an alcohol of Formula 10-8, following analogous procedures as described in Scheme 4.
- Intermediate compounds of Formula 10-7 can be prepared by oxidation of compounds of Formula 10-6 for example using Oxone in DMF.
- Oxone in DMF A person skilled in the art would understand that other suitable oxidizing agents and solvents may be used.
- Intermediate compounds of Formula 10-6 can be prepared by a metal-catalyzed cross-coupling reaction of a compound of Formula 10-5 with a boronic acid such as a substituted phenyl boronic acid of Formula 1-7, in which X 1 is chloro, in the presence of a palladium catalyst such as dichloro [ ⁇ , ⁇ bis(di-tert-butylphosphino)]ferrocene palladium (II), a base such as potassium phosphate tribasic in an inert solvent such as 1,4-dioxane with heating at elevated temperatures either thermally or with microwave irradiation.
- a palladium catalyst such as dichloro [ ⁇ , ⁇ bis(di-tert-butylphosphino)]ferrocene palladium (II)
- a base such as potassium phosphate tribasic in an inert solvent such as 1,4-dioxane with heating at elevated temperatures either thermally or with microwave irradiation.
- Intermediate compounds of Formula 10-5 can be prepared by a lithium-halogen exchange of a compound of Formula 10-3 with an alkyllithium reagent (such as butyllithium) followed by reaction with a formylating reagent such as DMF, or N-formylmorpholine or ethyl formate in a suitable solvent such as THF, at a temperature from about -78 °C to 40 °C.
- an alkyllithium reagent such as butyllithium
- a formylating reagent such as DMF, or N-formylmorpholine or ethyl formate
- THF suitable solvent
- Compounds of Formula 10-3 can in turn be prepared by a methylation reaction of a compound of Formula 10-2 in the presence of a base such as potassium carbonate, suitable methylating agents include iodomethane, dimethylsulfate, methyl triflate, methyl mesylate in a suitable solvent such as THF, at a temperature from about -20 °C to 60 °C. In the course of this reaction regioisomer 10-4 may also be formed.
- suitable methylating agents, bases and solvents may be used.
- Intermediate compounds of Formula 10-2 can be prepared by halogenation of a triazole of Formula 10-1.
- appropriate halogenating agents include N- bromosuccinimide and Br 2 ;
- X is iodine, appropriate halogenating agents include N- iodosuccinimide and iodine;
- X is chlorine, appropriate reagents include N- chlorosuccinimide and Cl 2 .
- the reaction takes place in a suitable solvent such as
- the invention also relates to those forms of the process in which a compound obtainable as an intermediate at any stage of the process is used as starting material and the remaining process steps are carried out, or in which a starting material is formed under the reaction conditions or is used in the form of a derivative, for example in a protected form or in the form of a salt, or a compound obtainable by the process according to the invention is produced under the process conditions and processed further in situ.
- Compounds of the invention and intermediates can also be converted into each other according to methods generally known to those skilled in the art. Intermediates and final products can be worked up and/or purified according to standard methods, e.g. using chromatographic methods, distribution methods, (re-) crystallization, and the like.
- protecting group a readily removable group that is not a constituent of the particular desired end product of the compounds of the present invention.
- the protection of functional groups by such protecting groups, the protecting groups themselves, and their cleavage reactions are described for example in standard reference works, such as J. F. W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973; in T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999; in “The Peptides”; Volume 3 (editors: E. Gross and J.
- Carbohydrates Monosaccharides and Derivatives), Georg Thieme Verlag, Stuttgart 1974; and in subsequent versions thereof.
- a characteristic of protecting groups is that they can be removed readily (i.e. without the occurrence of undesired secondary reactions) for example by solvolysis, reduction, photolysis or alternatively under physiological conditions (e.g. by enzymatic cleavage).
- mixtures of isomers that are formed can be separated into the individual isomers, for example diastereoisomers or enantiomers, or into any desired mixtures of isomers, for example racemates or mixtures of diastereoisomers.
- Mixtures of isomers obtainable according to the invention can be separated in a manner known to those skilled in the art into the individual isomers; diastereoisomers can be separated, for example, by partitioning between polyphasic solvent mixtures, recrystallisation and/or chromatographic separation, for example over silica gel or by e.g.
- medium pressure liquid chromatography over a reversed phase column and racemates can be separated, for example, by the formation of salts with optically pure salt-forming reagents and separation of the mixture of diastereoisomers so obtainable, for example by means of fractional crystallisation, or by chromatography over optically active column materials.
- solvents from which those solvents that are suitable for any particular reaction may be selected include those mentioned specifically or, for example, water, esters, such as lower alkyl-lower alkanoates, for example ethyl acetate, ethers, such as aliphatic ethers, for example diethyl ether, or cyclic ethers, for example tetrahydrofuran or dioxane, liquid aromatic hydrocarbons, such as benzene or toluene, alcohols, such as methanol, ethanol or 1- or 2- propanol, nitriles, such as acetonitrile, halogenated hydrocarbons, such as methylene chloride or chloroform, acid amides, such as dimethylformamide or dimethyl acetamide, bases, such as heterocyclic nitrogen bases, for example pyridine or N-methylpyrrolidin-2-one, carboxylic acid anhydrides, such as lower alkanoic acid anhydrides, for example acetic an
- the compounds of the present invention are either obtained in the free form, as a salt thereof, or as prodrug derivatives thereof.
- the compounds of the present invention may also form internal salts, e.g., zwitterionic molecules.
- the compounds of the present invention are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto.
- the terms “salt” or “salts” refers to an acid addition or base addition salt of a compound of the invention.
- Salts include in particular "pharmaceutical acceptable salts”.
- pharmaceutically acceptable salts refers to salts that retain the biological effectiveness and properties of the compounds of this invention and, which typically are not biologically or otherwise undesirable.
- Salts of compounds of the present invention having at least one salt-forming group may be prepared in a manner known to those skilled in the art.
- salts of compounds of the present invention having acid groups may be formed, for example, by treating the compounds with metal compounds, such as alkali metal salts of suitable organic carboxylic acids, e.g. the sodium salt of 2-ethylhexanoic acid, with organic alkali metal or alkaline earth metal compounds, such as the corresponding hydroxides, carbonates or hydrogen carbonates, such as sodium or potassium hydroxide, carbonate or hydrogen carbonate, with corresponding calcium compounds or with ammonia or a suitable organic amine, stoichiometric amounts or only a small excess of the salt-forming agent preferably being used.
- metal compounds such as alkali metal salts of suitable organic carboxylic acids, e.g. the sodium salt of 2-ethylhexanoic acid
- organic alkali metal or alkaline earth metal compounds such as the corresponding hydroxides, carbonates or hydrogen carbonates
- Acid addition salts of compounds of the present invention are obtained in customary manner, e.g. by treating the compounds with an acid or a suitable anion exchange reagent.
- Internal salts of compounds of the present invention containing acid and basic salt- forming groups e.g. a free carboxy group and a free amino group, may be formed, e.g. by the neutralisation of salts, such as acid addition salts, to the isoelectric point, e.g. with weak bases, or by treatment with ion exchangers.
- Salts can be converted into the free compounds in accordance with methods known to those skilled in the art.
- Metal and ammonium salts can be converted, for example, by treatment with suitable acids, and acid addition salts, for example, by treatment with a suitable basic agent.
- Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids, e.g., acetate, aspartate, benzoate, besylate, bromide/hydrobromide, bicarbonate/carbonate, bisulfate/sulfate, camphorsulfonate, chloride/hydrochloride,
- Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric
- Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, sulfosalicylic acid, and the like.
- Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
- Inorganic bases from which salts can be derived include, for example, ammonium salts and metals from columns I to XII of the periodic table.
- the salts are derived from sodium, potassium, ammonium, calcium, magnesium, iron, silver, zinc, and copper; particularly suitable salts include ammonium, potassium, sodium, calcium and magnesium salts.
- Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like.
- Certain organic amines include isopropylamine, benzathine, cholinate, diethanolamine, diethylamine, lysine, meglumine, piperazine and tromethamine.
- adipic acid adipate
- L- ascorbic acid ascorbate
- capric acid caprate
- sebacic acid sebacate
- xinafoate L-glutamic acid
- glutaric acid glutaric acid
- trifenatate triphenylacetic acid
- galactaric acid/mucic acid galactaric acid/mucic acid
- the pharmaceutically acceptable salts of the present invention can be synthesized from a parent compound, a basic or acidic moiety, by conventional chemical methods.
- such salts can be prepared by reacting free acid forms of these compounds with a stoichiometric amount of the appropriate base (such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like), or by reacting free base forms of these compounds with a stoichiometric amount of the appropriate acid.
- a stoichiometric amount of the appropriate base such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like
- Such reactions are typically carried out in water or in an organic solvent, or in a mixture of the two.
- use of non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile is desirable, where practicable.
- Lists of additional suitable salts can be found, e.g., in "Remington's Pharmaceutical Sciences", 20th ed., Mack Publishing Company, Easton, Pa., (1985); and in "Handbook of Pharmaceutical Salts:
- the present invention also provides pro-drugs of the compounds of the present invention that converts in vivo to the compounds of the present invention.
- a pro-drug is an active or inactive compound that is modified chemically through in vivo physiological action, such as hydrolysis, metabolism and the like, into a compound of this invention following administration of the prodrug to a subject.
- the suitability and techniques involved in making and using pro-drugs are well known by those skilled in the art.
- Prodrugs can be conceptually divided into two non-exclusive categories, bioprecursor prodrugs and carrier prodrugs. See The Practice of Medicinal Chemistry, Ch. 31-32 (Ed. Wermuth, Academic Press, San Diego, Calif., 2001), and in subsequent versions thereof.
- bioprecursor prodrugs are compounds which are inactive or have low activity compared to the corresponding active drug compound; contain one or more protective groups, and are converted to an active form by metabolism or solvolysis. Both the active drug form and any released metabolic products should have acceptably low toxicity.
- Carrier prodrugs are drug compounds that contain a transport moiety, e.g. , that improve uptake and/or localized delivery to a site(s) of action.
- a transport moiety e.g.
- the linkage between the drug moiety and the transport moiety is a covalent bond
- the prodrug is inactive or less active than the drug compound
- any released transport moiety is acceptably non-toxic.
- the transport moiety is intended to enhance uptake, typically the release of the transport moiety should be rapid.
- it is desirable to utilize a moiety that provides slow release e.g. , certain polymers or other moieties, such as cyclodextrins.
- Carrier prodrugs can, for example, be used to improve one or more of the following properties: increased lipophilicity, increased duration of pharmacological effects, increased site-specificity, decreased toxicity and adverse reactions, and/or improvement in drug formulation (e.g. , stability, water solubility, suppression of an undesirable organoleptic or physiochemical property).
- lipophilicity can be increased by esterification of (a) hydroxyl groups with lipophilic carboxylic acids (e.g., a carboxylic acid having at least one lipophilic moiety), or (b) carboxylic acid groups with lipophilic alcohols (e.g., an alcohol having at least one lipophilic moiety, for example aliphatic alcohols).
- Exemplary prodrugs are, e.g. , esters of free carboxylic acids and 5-acyl derivatives of thiols and O-acyl derivatives of alcohols or phenols, wherein acyl has a meaning as defined herein.
- Suitable prodrugs are often pharmaceutically acceptable ester derivatives convertible by solvolysis under physiological conditions to the parent carboxylic acid, e.g.
- lower alkyl esters lower alkyl esters, cycloalkyl esters, lower alkenyl esters, benzyl esters, mono- or di-substituted lower alkyl esters, such as the co-(amino, mono- or di-lower alkylamino, carboxy, lower alkoxycarbonyl)-lower alkyl esters, the oc-(lower alkanoyloxy, lower alkoxycarbonyl or di-lower alkylaminocarbonyl)- lower alkyl esters, such as the pivaloyloxymethyl ester and the like conventionally used in the art.
- amines have been masked as arylcarbonyloxymethyl substituted derivatives which are cleaved by esterases in vivo releasing the free drug and formaldehyde (Bundgaard, /. Med. Chem. 2503 (1989)).
- drugs containing an acidic NH group such as imidazole, imide, indole and the like, have been masked with N-acyloxymethyl groups (Bundgaard, Design of Prodrugs, Elsevier (1985)), and in subsequent versions thereof. Hydroxy groups have been masked as esters and ethers.
- EP 039,051 (Sloan and Little) discloses Mannich-base hydroxamic acid prodrugs, their preparation and use.
- the compounds of the present invention may also be obtained in the form of hydrates, or their crystals may, for example, include the solvent used for crystallization. Different crystalline forms may be present.
- the compounds of the present invention may inherently or by design form solvates with pharmaceutically acceptable solvents (including water); therefore, it is intended that the invention embrace both solvated and unsolvated forms.
- solvate refers to a molecular complex of a compound of the present invention (including pharmaceutically acceptable salts thereof) with one or more solvent molecules.
- solvent molecules are those commonly used in the pharmaceutical art, which are known to be innocuous to the recipient, e.g., water, ethanol, and the like.
- hydrate refers to the complex where the solvent molecule is water.
- the compounds of the present invention, including salts, hydrates and solvates thereof may inherently or by design form polymorphs.
- Compounds of the invention in unoxidized form may be prepared from N-oxides of compounds of the invention by treating with a reducing agent (e.g., sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride, phosphorus trichloride, tribromide, or the like) in a suitable inert organic solvent (e.g. acetonitrile, ethanol, aqueous dioxane, or the like) at 0 to 80°C.
- a reducing agent e.g., sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride, phosphorus trichloride, tribromide, or the like
- a suitable inert organic solvent e.g. acetonitrile, ethanol, aqueous dioxane, or the like
- UV Diode Array at 214nm - 400nm
- reaction mixture was then cooled to rt and (R) -2- chloro-a-methyl benzyl alcohol (3.0 g, 19.1 mmol) was added.
- the newly formed reaction mixture was refluxed for lh and then cooled to rt.
- the mixture was filtered and the filtrate was diluted with water (50 ml), and extracted with dichloromethane (3x 75 mL). The combined organic phases were washed with brine, dried over sodium sulfate, filtered, and concentrated.
- the reaction was treated with 2M NaOH to pHIO, 10% sodium thiosulfate (2 mL) was added and the mixture partitioned between ethyl acetate and water.
- the aqueous phase was then acidified with citric acid and extracted with ethyl acetate.
- the organic phase was then dried over magnesium sulfate, filtered and concentrated in vacuo, to provide the title compound.
- 4-(4-Chlorophenyl)-l-methyl-lH-pyrazol-5-amine was prepared from 4-bromo-l- methyl-lH-pyrazol-5-amine and (4-chlorophenyl)boronic acid following analogous procedures as described in Intermediate 8, using dichloro [ ⁇ , bis(di-tert-butylphosphino)]ferrocene palladium (II) as catalyst, potassium phosphate as base, and dioxane/water as solvent, at 120 °C in a sealed reaction vessel, with a reaction time of 1 h.
- Ethyl 1 - (4'-(5 -amino- 1 -methyl- 1 H-pyrazol-4-yl) - [ 1 , 1 '-biphenyl] -4- yl)cyclopropanecarboxylate was prepared from 4-(4-chlorophenyl)-l -methyl- lH-pyrazol-5- amine (INT-9) and ethyl l-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl)cyclopropanecarboxylate following the procedure in Intermediate 9, with a reaction temperature of 100 °C.
- OXONE® 275 mg, 0.447 mmol was added to a solution of 4-(4-chlorophenyl)-l- methyl-lH-l,2,3-triazole-5-carbaldehyde (90 mg, 0.406 mmol) in DMF (5 mL) and the resulting suspension was stirred at room temperature overnight.
- HC1 1M 10 mL was added and the resulting mixture was extracted with ethyl acetate (3x10 mL).
- Tetrahydro-2H-pyran-4-carbaldehyde 500mg, 4.38mmol was placed in a vial with anhydrous THF (5mL) and cooled on ice. Methylmagnesium bromide (3M in diethyl ether) (2.19mL, 6.57mmol) was slowly added (exothermic). A white precipitate formed. The reaction mixture was stirred at room temperature for 1 hour and quenched with saturated ammonium chloride (3mL) (slowly at first - exothermic) and water (3mL). The supernatant organic phase was passed through a phase separator filled with dry MgS0 4 (5-10g).
- Example 3-2 was prepared following analogous procedures as described in Example
- dichloropalladium(II) (7.28 mg, 0.011 mmol), were suspended in dioxane (1 mL) and water (0.25 mL) and degassed and flushed with nitrogen. The mixture was sealed and heated at 100°C for 30 minutes by microwave irradiation. The reaction mixture was treated with 10% aqueous citric acid to pH 5-6 and filtered through a pad of celite. The filtrate was partitioned between ethyl acetate and water. The aqueous phase was extracted further with ethyl acetate, and the combined organic layers were dried over magnesium sulfate, filtered and concentrated in vacuo.
- (R)-l-phenylethyl 4-(4-hydroxvphenvl)-l-methyl-lH-pyrazol-5-ylcarbamate A mixture of (R)-l-phenylethyl 4-bromo-l -methyl- lH-pyrazol-5-ylcarbamate (INT-3b, 200 mg, 0.62 mmol), 4-hydroxyphenylboronic acid (128 mg, 0.93 mmol), NaHCC>3 (156 mg, 1.86 mmol), Pd(dppf)Cl2 (51 mg, 0.062 mmol), dioxane ( 2 mL), and water (2 mL) was heated under nitrogen with stirring at 100 °C for 3 hours.
- racemate compound (6 racemate) was prepared from the racemate reactants following similar procedures. HPLC-MS calculated for C29H2 8 N 3 O4 (M+H) 482.2, found 482.2; R T 1.24 min. (LCMS Method 2).
- a suitable solvent such as ethyl acetate, 3 -methyl- 1-butanol or 4-methyl-2-pentanone (7 mL), resulting in a slurry which was stirred at room temperature for approximately 72 hours. The solvent was then removed, and the crystalline salts were collected and dried in a vacuum oven at 40 °C for 24 hours.
- crystalline (R)-l-(4'-(l-methyl-5-((l- phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid potassium salt is characterized as having an XRPD diffraction pattern substantially the same as Figure 1; a DSC thermogram substantially the same as Figure 2A, a TGA thermogram substantially the same as Figure 2A; or combinations thereof.
- the mixture was stirred and heated at 55 °C to dissolve visible solids; subsequently, 50 ⁇ lL of a microemulsion preconcentrate (e.g., percentage by weight: 16.7% ethanol, 8.3% propylene glycol, 16.9% LABRAFIL®M2125CS and 58.1% CREMOPHOR EL®) was added to the mixture, which was stirred to give a clear solution with a pH of -8.5.
- a microemulsion preconcentrate e.g., percentage by weight: 16.7% ethanol, 8.3% propylene glycol, 16.9% LABRAFIL®M2125CS and 58.1% CREMOPHOR EL®
- the isomeric mixture was subjected to chiral HPLC conditions (Column: 21x25mm Whelk O-l; Solvents: Hexane/MeOH/EtOH (7:2:1) with 1% HO Ac; Flow Rate: 1 mL/min; Temperature: ambient; Run Time: 25 mins) to give: (i) Isomer A (Retention time: 17.08 mins); and (ii) Isomer B
- Example 10-2 was prepared following the procedures in Example 10-1, substituting appropriate reagents where required.
- the isomeric mixture was subjected to chiral SFC conditions (Column: Chiralcel OJ-H 250 x 10 mm, 5 ⁇ ; Mobile phase: 50% methanol / 50% C0 2 ; Flow rate: 10 mL/min; Detection: UV @ 220 nm) to give: (i) Isomer A (SFC Retention Time: 2.78 mins), LCMS R T 4.02min; MS m/z 483.1 [M+H] + (LCMS method 3); and (ii) Isomer B (SFC Retention Time: 4.34 min). LCMS R T 4.11min; MS m/z 483.3 [M+H] + (LCMS method 3).
- Examples 18-2 and 18-3 were prepared following the procedures in Example 18-1 substituting appropriate reagents where required.
- Methyl l-(4'-(l-methyl-5-((l-phenylbut-2-ynyloxy)carbonylamino)-lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylate was prepared as a yellow foam following procedures described in Example 19, step 1, with l-phenylbut-2-yn-l-ol (INT-22) as reactant. Rt 1.31min; MS m/z 520.4 [M+H]+; Method 2.
- Lithium hydroxide monohydrate (9.69 mg, 0.231 mmol) was added to a solution of methyl l-(4'-(l-methyl-5-((l-phenylbut-2-ynyloxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylate (30 mg, 0.058 mmol) in a mixture of THF (0.5 mL) and water (0.500 mL) and the resulting mixture stirred overnight. The reaction was acidified with acetic acid, diluted with water (5 mL) and extracted with ethyl acetate (3x10 mL).
- Ethyl l-(4'-(5-((3-methoxy-l-phenylpropoxy)carbonylamino)-l-methyl-lH-pyrazol- 4-yl)biphenyl-4-yl)cyclopropanecarboxylate was prepared as a yellow foam following the procedures described in Example 19 step 1, with 3-methoxy-l-phenylpropan-l-ol as reactant. (M+H 554.5).
- the reaction was acidified using 1M HC1 solution and the resulting mixture extracted with ethyl acetate (3x10 mL). The combined organic solutions were washed with brine (5 mL), dried over sodium sulphate, filtered and evaporated. The residue was dissolved in DCM (10 mL) and MP-isocyanate resin (200 mg 0.298 mmol, 1.49 mmol/g) was added. The resulting mixture was stirred overnight at room temperature. The resin was removed by filtration, washed with DCM (3x5 mL) and the combined washings and filtrate concentrated in vacuo.
- Ethyl l-(4'-(5-((cyclopropyl(phenyl)methoxy)carbonylamino)-l-methyl-lH-pyrazol- 4-yl)biphenyl-4-yl)cyclopropanecarboxylate was prepared as a yellow foam following the procedures described in Example 19 step 1, with (+)cyclopropyl(phenyl)methanol as reactant. (M+H 536.4).
- Lithium hydroxide monohydrate (61.4 mg, 1.463 mmol) was added to a solution of ethyl l-(4'-(5-((cyclopropyl(phenyl)methoxy)carbonylamino)-l-methyl-lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylate (135 mg, 0.244 mmol) in a mixture of water (1.0 mL) and THF (1 mL) and the resulting mixture stirred at room temperature for 90 min. The reaction was heated to 50°C for 30 hrs.
- Ethyl 1 -(4'-(5-((2-ethoxy- 1 -phenylethoxy)carbonylamino)- 1 -methyl- lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylate was prepared as a yellow foam following procedures described in Example 19 step 1, with 2-ethoxy-l-phenylethanol (INT-24) as reactant. (M+H 554.5)
Abstract
The present invention relates to compounds of Formula (1), or pharmaceutically acceptable salts thereof and their pharmaceutical compositions, wherein variables are as defined herein, which are useful as modulators of the activity of lysophosphatidic acid (LPA).
Description
COMPOSITIONS AND METHODS FOR MODULATING LPA RECEPTORS
Cross-Reference to Related Applications
[0001] This application claims the benefit of U.S. provisional application serial number 61/472,547 filed April 6, 2011 ; and of U.S. provisional application serial number 61/607,393 filed March 6, 2012; each of which is incorporated herein by reference in their entirety.
Technical Field
[0002] The present invention relates to compositions and methods for modulating the activity of lysophosphatadic acid (LPA receptors).
Background
[0003] Lysophosphatidic acid (LPA) is a bioactive phospholipid with growth factor- like properties regulating cell motility, proliferation, survival, and differentiation. LPA acts on a number of G-protein coupled receptors, including LPAl, LPA2, and LP A3. Several other receptors have been reported to respond to LPA (GPR23, GPR92, GPR87, P2Y5, P2Y10) (Noguchi et al., Curr. Opin. Pharmacol. 2009). LPA receptors have broad and overlapping expression patterns (Choi et al., Annu. Rev. Pharmacol. Toxicol. 2010). LPAl is expressed in all human tissues examined with the exception of liver and leukocytes. LPA2 and LP A3 receptors are also broadly expressed. LPA receptors have been shown to couple to Gj/0, Gq/n, and G12/13 pathways, and affect multiple signaling pathways inside the cell. LPA is primarily generated by the enzyme autotoxin (ATX) from the precursor lysophosphatidylcholine (Tokumura et al., /. Biol. Chem. 2002). ATX knockout mice die embryonically; heterozygote mice survive to adulthood with about half of plasma LPA level as of wild-type mice (Tanaka et al., /. Biol. Chem. 2006). Enhanced secretion of LPA has been associated with fibrosis, wound healing, inflammation, and cancer (Choi et al., Annu. Rev. Pharmacol. Toxicol. 2010).
[0004] Several studies have shown that LPAl plays an important role in the initiation of fibrotic disorders. LPA concentration is significantly increased in bronchoalveolar lavage fluid (BAL) of bleomycin-challenged mice, and bleomycin-treated LPAl knockout mice had a lower incidence of lung fibrosis and better survival compared to wild-type animals (Tager et al., Nat. Med. 2008). LPAl mediates vascular leakage and increases fibroblast recruitment. LPAl is the most highly expressed LPA receptor in fibroblasts isolated from patients with idiopathic
pulmonary fibrosis (IPF) (Tager et al., Nat. Med. 2008). BAL obtained from IPF patients has a high LPA level and it mediates chemo taxis of human fetal lung fibroblasts, and such effects could be blocked by the LPA1-3 receptor antagonist Kil6425 (Tager et al., Nat. Med. 2008). LPA signaling might also play a role in airway inflammation and remodeling as well (Zhao et al., Cell Signal. 2009).
[0005] Idiopathic pulmonary fibrosis (IPF) and other fibrotic lung diseases are associated with high morbidity and mortality, and are generally refractory to currently available pharmacological therapies. Although considerable progress has been made in diagnosing the disease, no effective treatments are known. Lung transplantation is currently the only therapeutic option available. Thus, there is a clear unmet need for better treatments.
[0006] LPA was also implicated in the development of renal fibrosis that was not induced by bleomycin, suggesting that LPA signaling is likely involved in fibrosis in different organs initiated by different underlying causes. In the unilateral ureteral obstruction (UUO) kidney fibrosis model, the development of renal fibrosis was significantly attenuated in LPA1 knockout mice (Pradere et. al., /. Am. Soc. Nephrol. 2007).
[0007] A selective or pan- LPA receptor antagonist might have utility towards other disease indications. A large amount of literature has linked expression of ATX and LPA receptor(s) with increased tumorigenesis, invasion, and metastases, in particular for ovarian cancer, breast cancer and their metastases (Liu et al., Cancer Cell 2009; Boucharaba et al., Proc. Natl. Acad. Sci. U. S. A. 2006). LPA signaling has also been implicated in rheumatoid arthritis (Bourgoin & Zhao, Curr. Opin. Investig. Drugs. 2010).
Disclosure of the Invention
[0008] The present invention relates to compositions and methods for modulating the activity of lysophosphatadic acid (LPA receptors). In one aspect, the present invention relates to compounds which act as inhibitors of LPA, and methods of using such compounds to treat a disease or condition associated with one or more LPA receptors.
[0009] In a first embodiment, the invention provides a compound having Formula (1):
 nyl, phenyl fused to a cyclopentyl, C3-7 cycloalkyl, spiro[2,5]octanyl, or
nyl, phenyl fused to a cyclopentyl, C3-7 cycloalkyl, spiro[2,5]octanyl, or
 C3 of the pyridyl ring is attached to -(CR1R2)P-R3;
C3 of the pyridyl ring is attached to -(CR1R2)P-R3;
Z1 and Z2 are independently CH or N; or Z2 is C if attached to R4a;
R1 is hydrogen, halo, hydroxy or C02H;
R2 is hydrogen, halo or methyl; or
R1 and R2 together form cyclopropyl, or a monocyclic 4-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O and S; wherein said 4-6 membered ring is unsubstituted or substituted by 1-2 R9 groups;
R3 is -CO2H, -C(0)NR-(CRR')o-2-CN, -C(0)NRS02R10 or -NR-C(0)R10;
R4a and R4b are independently hydrogen, halo, Ci_6 alkyl, Ci_6 alkoxy or Ci_6 hydroxyalkyl;
R5 is methyl or ethyl;
R6 is hydrogen, Ci_6 alkyl, -(CRR')i_2-0(Ci_4 alkyl), C2^ alkynyl or cyclopropyl;
R6a is hydrogen or Ci_6 alkyl;
R6b is Ci_6 alkyl; cyclopropyl; phenyl substituted with R7 and R8; or a 5-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O or S;
R7 and R8 are independently hydrogen, Ci^ alkyl or halo;
R9 is -C(0)Rn, -C(0)ORn or S02Rn;
R10 and R11 are independently Ci^ alkyl;
R and R' are independently hydrogen or Ci_6 alkyl;
m and n are independently 1-4; and
p is 0-2; or
a stereoisomer or pharmaceutically acceptable salt thereof.
[0010] In a second embodiment, the invention provides a compound of Formula (1) or pharmaceutical acceptable salt thereof, wherein R6b is isopropyl; methyl; cyclopropyl; phenyl substituted with R7 and R8; tetrahydrofuranyl or tetrahydropyranyl; and the other substituents are as defined above.
[0011] In a third embodiment, the invention provides a compound having Formula (2):

[0012] In a fourth embodiment, R1 and R2 in any of the above embodiments are
independently hydrogen or halo; more particularly, R1 and R2 are independently hydrogen or fluoro. In embodiments where p=2, R1 and R2 in each -(CR^2)- repeating group are defined independently, and encompass identical and non-identical -(CR^2)- repeating groups.
Alternatively, R1 and R2 together form cyclopropyl, oxetanyl, tetrahydropyanyl or piperidinyl substituted with 1-2 R9 groups; wherein R9 is as defined in Formula (1); more particularly, R1 and R2 together form cyclopropyl.
[0013] In any of the above embodiments, R6 can be hydrogen, methyl, methoxymethylene, methoxyethylene, ethoxymethylene, isopropoxymethylene, prop-l-ynyl or cyclopropyl; more particularly, R6 is hydrogen or methyl.
[0014] In a fifth embodiment, R5 in any of the above embodiments is methyl; and R6 is hydrogen or methyl; more particularly, R5 and R6 are methyl.
[0015] In any of the above embodiments, Z2 can be CH or C if attached to R4a; and R4a is hydrogen, halo, Ci_6 alkyl, Ci_6 alkoxy or Ci_6 hydroxy alkyl; more particularly, R4a is hydrogen, fluoro, methyl, methoxy or hydroxymethyl. In other embodiments, Z2 is N and R4a is hydrogen.
[0016] In any of the above embodiments, B can be phenyl unsubstituted or substituted by
halo or Ci_6 alkyl; phenyl fused to a cyclopentyl; cyclohexyl; spiro[2,5]octanyl, or
 C3 of the pyridyl ring is attached to -(CR1R2)P-R3. In particular embodiments, B is phenyl unsubstituted or substituted by R4b wherein R4b is hydrogen, halo or Ci-6 alkyl; more particularly, R4b is hydrogen, fluoro, chloro or methyl.
C3 of the pyridyl ring is attached to -(CR1R2)P-R3. In particular embodiments, B is phenyl unsubstituted or substituted by R4b wherein R4b is hydrogen, halo or Ci-6 alkyl; more particularly, R4b is hydrogen, fluoro, chloro or methyl.
[0017] In a sixth embodiment, the invention provides a compound having Formula (3):

R1 and R2 together form cyclopropyl;
R3, R4a, R4b, R7, R8, m, n and p are as defined in Formula (1); or
a stereoisomer or pharmaceutically acceptable salt thereof.
[0018] In a seventh embodiment, the invention provides a compound of Formula (4):

or a stereoisomer or pharmaceutically acceptable salt thereof; wherein the substituents are as defined above.
[0019] In an eighth embodiment, R a is hydrogen, halo, Ci_6 alkyl, Ci_6 alkoxy or Ci_6 hydroxyalkyl; and R4b is hydrogen, halo or Ci_6 alkyl. In particular embodiments, R4a is hydrogen, fluoro, methyl, methoxy or hydroxymethyl; and R4b is hydrogen, fluoro, chloro or methyl. In other embodiments, R4a is hydrogen, halo, Ci_6 alkyl or Ci_6 hydroxyalkyl; more particularly, R4a is hydrogen, fluoro, methyl or hydroxymethyl.
[0020] In a ninth embodiment, m=l and n=l in any of the above embodiments; and R4a and R4b are hydrogen.
[0021] In a tenth embodiment, R3 in any of the above embodiment is -CO2H.
[0022] In an eleventh embodiment, R7 and R8 in any of the above embodiments are independently hydrogen, halo or methyl; and more particularly, hydrogen, fluoro or chloro.
[0023] In a twelfth embodiment, the invention provides a compound selected from:
2-(4-{4-[5-({ [l-(2-chlorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid (la);
2-(4- { 4- [5 -( { [( 1 R)- 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yl]phenyl}phenyl)acetic acid (lb);
2-(4-{4-[5-({ [l-(2-chlorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2-(4- { 4- [5 -( { [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -ethyl- 1 H-pyrazol-4- yl]phenyl}phenyl)acetic acid (2-1);
4-{4-[5-({ [(lR)-l-(2-chlorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH-pyrazol-4- yl]phenyl} benzoic acid (2-2);
4-{4-[5-({ [l-(2-chlorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH-pyrazol-4- yl]phenyl} benzoic acid;
2-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid (2-3);
2-(4- {4-[ 1 -methyl-5-( { [ l-phenylethoxy]carbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2- { 4- [4- ( 1 -methyl- 5 - { [( 1 -pheny lpropoxy )carbonyl] amino } - 1 H-pyrazol-4- yl)phenyl]phenyl} acetic acid (2-4);
2-(4-{4-[l-methyl-5-({ [l-(2-methylphenyl)ethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl} pheny l)acetic acid (2-5);
2- { 4- [4-(5 - { [( 1 -cyclopropylethoxy)carbonyl] amino } - 1 -methyl- lH-pyrazol-4-
yl)phenyl]phenyl} acetic acid (2-6);
2-(4-{4-[5-({ [(l R)- 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yl]phenyl}cyclohexyl)acetic acid (3-1);
2-(4- { 4- [5 -( { [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yljphenyl } cyclohexyl) acetic acid;
2-(4-{4-[5-({ [(lR)-l-(2-chloro-4-fluorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH- pyrazol-4-yl]phenyl}cyclohexyl)acetic acid (3-2);
2- (4-{4-[5-({ [l-(2-chloro-4-fluorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH- pyrazol-4-yl]phenyl } cyclohexyl) acetic acid;
3- (4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl} amino)- lH-pyrazol-4- yl]phenyl}phenyl)oxetane-3-carboxylic acid (4-1);
3- (4- { 4- [ l-methyl-5-( { [ l-phenylethoxy]carbonyl } amino)- lH-pyrazol-4- yl]phenyl}phenyl)oxetane-3-carboxylic acid;
1- [(tert-butoxy)carbonyl]-4-(4- { 4-[ 1 -methyl-5-( { [( 1R)- 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4-yl]phenyl }phenyl)piperidine-4-carboxylic acid (4-2);
1- [(tert-butoxy)carbonyl]-4-(4- { 4-[ 1 -methyl-5-( { [ 1 -phenylethoxyjcarbonyl } amino)- 1H- pyrazol-4-yl]phenyl}phenyl)piperidine-4-carboxylic acid;
4- (4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)oxane-4-carboxylic acid (4-3);
4-(4- { 4- [ l-methyl-5-( { [ l-phenylethoxy]carbonyl } amino)- lH-pyrazol-4- yljphenyl }phenyl)oxane-4-carboxylic acid;
( 1 R)- 1 -phenylethyl N- [4-(4- { 4- [(methanesulfonylcarbamoyl)methyl]phenyl }phenyl)- 1 - methyl- lH-pyrazol-5-yl]carbamate (5);
1-phenylethyl N- [4-(4- { 4-[(methanesulfonylcarbamoyl)methyl]phenyl }phenyl)- 1 - methyl- 1 H-pyrazol- 5 -yl] carbamate ;
(R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid (6a);
l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid (6a racemic);
l-(4- { 4- [5-( { [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l-carboxylic acid (7-1);
1- (4-{2-fluoro-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l-carboxylic acid (7-2);
1 -(4- { 2-fluoro-4- [ 1 -methyl-5-( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
2- (4-{4-[5-({ [(l R)- 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yl]phenyl}phenyl)-2,2-difluoroacetic acid (7-3);
2- (4-{4-[5-({ [l-(2-chlorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH-pyrazol-4- yl]phenyl}phenyl)-2,2-difluoroacetic acid;
1 -(4- { 3 -methyl-4- [ 1 -methyl-5 -( { [( 1R)- 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (7-4);
1 -(4- { 3 -methyl-4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
3- (4-{4-[5-({ [(l R)- 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yl]phenyl}phenyl)propanoic acid (8);
3 -(4- { 4- [5 -( { [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yl]phenyl}phenyl)propanoic acid;
1- (2-fluoro-4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (9-1);
1 -(2-fluoro-4- { 4- [ 1 -methyl-5-( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
2- (2-fluoro-4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid (9-2);
2-(2-fluoro-4-{4-[l-methyl-5-({ [l-phenylethoxy]carbonyl} amino)- lH-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2-(2-chloro-4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid (9-3);
2-(2-chloro-4- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2-(2-methyl-4- { 4- [ 1 -methyl-5 -( { [( 1R)- 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4- yl]phenyl}phenyl)acetic acid (9-4);
2-(2-methyl-4- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2,2-difluoro-2-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl} amino)- lH-pyrazol- 4-yl]phenyl}phenyl)acetic acid (9-5);
2,2-difluoro-2-(4- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxy] carbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)acetic acid;
l-(4-{3-fluoro-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l-carboxylic acid (9-6);
1 -(4- { 3 -fluoro-4- [ 1 -methyl-5-( { [ 1 -phenylethoxy] carbonyl } amino)- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 -(4- { 2-methyl-4- [ 1 -methyl-5 -( { [( 1R)- 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (9-7);
1 -(4- { 2-methyl-4- [ 1 -methyl-5 -( { [ 1 -phenylethoxy] carbonyl } amino)- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
3-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)propanoic acid (9-8);
3-(4-{4-[l-methyl-5-({ [l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)propanoic acid;
l-(4-{3-methoxy-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol- 4-yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (9-9);
1 -(4- { 3-methoxy-4-[ 1 -methyl-5-({ [ 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{2-methoxy-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol- 4-yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (9-10);
1 -(4- {2-methoxy-4-[ 1 -methyl-5-({ [ 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 -methyl- 5 - { 4- [ 1 -methyl- 5 - ( { [( 1 R)- 1 -phenylethoxy ] c arbonyl } amino) - 1 H-pyrazol-4- yl]phenyl}-2,3-dihydro-lH-indene-l-carboxylic acid (10-1);
1 -methyl- 5 - { 4- [ 1 -methyl- 5 - ( { [ 1 -phenylethoxy ] carbonyl } amino) - 1 H-pyrazol-4- yljphenyl} -2,3-dihydro- lH-indene- 1-carboxylic acid;
6- { 4- [5-({ [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- lH-pyrazol-4- yl]phenyl}-2,3-dihydro-lH-indene-l-carboxylic acid (10-2);
l-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}arnino)-lH-l,2,3-triazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (11);
l-(4-{4-[l-methyl-5-({ [l-phenylethoxy]carbonyl}amino)-lH-l,2,3-triazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{4-[l-methyl-5-({ [(lR)-l-phenylpropoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (12-1);
l-(4-{4-[l-methyl-5-({ [l-phenylpropoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 - { 4- [4- ( 1 -methyl- 5 - { [( 1 -pheny lbutoxy )carbonyl] amino } - 1 H-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (12-2);
1 - { 4- [4-(5 - { [(2,2-dimethyl- 1 -pheny lpropoxy)carbonyl] amino } - 1 -methyl- lH-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (12-3);
1 - (4- { 4- [ 1 -methyl- 5 - ( { [ 1 - (3 -methylphenyl)ethoxy] carbonyl } amino) - 1 H-pyrazol-4- yljphenyl} pheny l)cyclopropane-l -carboxylic acid (12-4);
l-(4-{4-[5-({ [(lR)-l-(2-chloro-4-fluorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH- pyrazol-4-yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (12-5);
1- (4-{4-[5-({ [l-(2-chloro-4-fluorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH- pyrazol-4-yl]phenyl}phenyl)cyclopropane-l -carboxylic acid;
1 - { 4- [4-(5 - { [(tert-butoxy)carbonyl] amino } - 1 -methyl- lH-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (12-6);
2- acetamido- 3 - (4- { 4- [ 1 -methyl- 5 - ( { [( 1 R) - 1 -phenylethoxy] carbonyl } amino)- 1 H-pyrazol- 4-yl]phenyl} pheny l)propanoic acid (13-1);
2- acetamido- 3 - (4- { 4- [ 1 -methyl- 5 - ( { [ 1 -phenylethoxy] c arbony 1 } amino)- 1 H-pyrazol-4- yljphenyl} pheny l)propanoic acid;
2-hydroxy-2-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl} pheny l)propanoic acid (13-2);
2-hydroxy-2-(4- {4-[ 1 -methyl-5-({ [ 1 -phenylethoxy] carbonyl } amino)- lH-pyrazol-4- yl]phenyl} pheny l)propanoic acid (13-2);
l-(4-{6-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl} amino)- lH-pyrazol-4-yl]pyridin- 3-yl}phenyl)cyclopropane-l-carboxylic acid (14);
l-(4-{6-[l-methyl-5-({ [l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4-yl]pyridin-3- yl }phenyl)cyclopropane- 1 -carboxylic acid;
l-{4-[2-(hydroxymethyl)-4-[l-methyl-5-({[(lR)-l-phenylethoxy]carbonyl}amino)-lH- pyrazol-4-yl]phenyl]phenyl } cyclopropane- 1 -carboxylic acid (15);
1- { 4- [2-(hydroxymethyl)-4- [ l-methyl-5-( { [ 1 -phenylethoxyjcarbonyl } amino)- 1H- pyrazol-4-yl]phenyl]phenyl } cyclopropane- 1-carboxy lie acid;
l-(6-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}pyridin-3-yl)cyclopropane- 1-carboxy lie acid (16);
l-(6-{4-[l-methyl-5-({ [l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}pyridin-3-yl)cyclopropane- 1-carboxy lie acid;
6-{4-[l-methyl-5-({[(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}spiro[2.5]octane-l-carboxylic acid (17);
6- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4- yl]phenyl}spiro[2.5]octane-l-carboxylic acid;
1 - { 4- [4-( 1 -methyl-5- { [( 1 -phenyl- 1 -deuteroethoxy)carbonyl] amino } - 1 H-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1-carboxy lie acid (18-1);
1 - { 4- [4- ( 1 -methyl- 5 - { [( 1 -phenyl- 1 -deutero- 1 - trideuteromethylmethoxy)carbonyl] amino } - 1 H-pyrazol-4-yl)phenyl]phenyl } cyclopropane- 1 - carboxylic acid (18-2);
1 - { 4- [4- ( 1 -methyl- 5 - { [( 1 -pentadeuterophenyl- 1 -deutero- 1 - trideuteromethylmethoxy)carbonyl] amino } - 1 H-pyrazol-4-yl)phenyl]phenyl } cyclopropane- 1 - carboxylic acid (18-3);
1- {4-[4-(5-{ [(2-methoxy- l-phenylethoxy)carbonyl]amino }- 1-methyl- lH-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (19);
l-(4-{4-[l-methyl-5-({ [(l-phenylbut-2-yn-l-yl)oxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (20);
1- {4-[4-(5- { [(3-methoxy- l-phenylpropoxy)carbonyl] amino } - 1-methyl- 1 H-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (21);
l-(4- { 4- [5-( { [cyclopropyl(phenyl)methoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (22);
1 - { 4- [4-(5- { [(2-ethoxy- 1 -phenylethoxy)carbonyl] amino } - 1 -methyl- 1 H-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (23);
l-[4-(4-{ l-methyl-5-[({[(2R)-3-methylbutan-2-yl]oxy}carbonyl)amino]-lH-pyrazol-4- yl}phenyl)phenyl]cyclopropane-l -carboxylic acid (24a);
l-[4-(4-{ l-methyl-5-[({[(2S)-3-methylbutan-2-yl]oxy}carbonyl)amino]-lH-pyrazol-4- yl}phenyl)phenyl]cyclopropane-l -carboxylic acid (24b);
l-[4-(4-{ l-methyl-5-[({[3-methylbutan-2-yl]oxy}carbonyl)amino]-lH-pyrazol-4- yl }phenyl)phenyl]cyclopropane- 1 -carboxylic acid;
1 - { 4- [4-(5 - { [( 1 -cyclopropylethoxy)carbonyl] amino } - 1 -methyl- lH-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (25);
l-(4-{4-[l-methyl-5-({ [l-(2-methylphenyl)ethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (26);
l-(4-{4-[l-methyl-5-({ [l-(oxan-4-yl)ethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (27);
l-(4-{4-[l-methyl-5-({ [l-(oxolan-3-yl)ethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (28);
1 - { 4- [4-(5- { [(benzyloxy)carbonyl] amino } - 1 -methyl- 1 H-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (29-1);
l-(4-{4-[l-methyl-5-({ [(2-methylphenyl)methoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (29-2);
1 - { 4- [4-( l-methyl-5- { [(oxan-4-ylmethoxy)carbonyl] amino } - 1 H-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (29-3);
l-{4-[4-(l-methyl-5-{ [(oxolan-3-ylmethoxy)carbonyl]amino}-lH-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (29-4);
l-[6-(4-{ l-methyl-5-[({[(2R)-3-methylbutan-2-yl]oxy}carbonyl)amino]-lH-pyrazol-4- yl}phenyl)pyridin-3-yl]cyclopropane-l-carboxylic acid (30-1);
1 - [6-(4- { 1 -methyl-5- [( { [3 -methylbutan-2-yl]oxy } carbonyl)amino] - 1 H-pyrazol-4- yl}phenyl)pyridin-3-yl]cyclopropane-l -carboxylic acid;
l-{6-[4-(5-{ [(l -cyclopropylethoxy)carbonyl] amino } - 1 -methyl- 1 H-pyrazol-4- yl)phenyl]pyridin-3-yl} cyclopropane- 1 -carboxylic acid (30-2);
l-(6-{4-[l-methyl-5-({ [l-(oxolan-3-yl)ethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}pyridin-3-yl)cyclopropane-l -carboxylic acid (30-3);
1 - { 6- [3 -methoxy-4-( 1 -methyl-5- { [( 1 -phenyl- 1 -deutero- 1 - trideuteromethylmethoxy)carbonyl]amino}-lH-pyrazol-4-yl)phenyl]pyridin-3-yl}cyclopropane- 1-carboxylic acid (31);
l-(6-(3-methoxy-4-(l-methyl-5-(((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4- yl)phenyl)pyridin-3-yl)cyclopropanecarboxylic acid;
1 -acetyl-4-(4- { 4- [ 1 -methyl-5 -( { [( 1 R)- 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)piperidine-4-carboxylic acid (32);
1 -acetyl-4-(4- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)piperidine-4-carboxylic acid;
1- methanesulfonyl-4-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH- pyrazol-4-yl]phenyl}phenyl)piperidine-4-carboxylic acid (33);
1 -methanesulfonyl-4-(4- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- 1H- pyrazol-4-yl]phenyl}phenyl)piperidine-4-carboxylic acid;
( 1 R)- 1 -phenylethyl N- { 4- [4-(4- { 1 - [(2-cyanoethyl)carbamoyl]cyclopropyl }
phenyl)phenyl]-l -methyl- lH-pyrazol-5-yl} carbamate (34); and
1 -phenylethyl N- { 4- [4-(4- { 1 - [(2-cyanoethyl)carbamoyl]cyclopropyl } phenyl)phenyl] - 1 - methyl- 1 H-pyrazol-5 -yl } carbamate; or
a stereoisomer or pharmaceutically acceptable salt thereof.
[0024] In another embodiment, the invention provides a compound selected from the group consisting of:
2- (4-{4-[5-({ [l-(2-chlorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid (la);
2-(4-{4-[5-({ [(lR)-l-(2-chlorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid (lb);
2-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid (2-3);
2-(4- { 4- [ l-methyl-5-( { [ l-phenylethoxy]carbonyl } amino)- lH-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2-(4-{4-[l-methyl-5-({ [l-(2-methylphenyl)ethoxy]carbonyl} amino)- lH-pyrazol-4- yl]phenyl}phenyl)acetic acid (2-5);
2-(4-{4-[5-({ [(lR)-l-(2-chlorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH-pyrazol-4- yl]phenyl}phenyl)-2,2-difluoroacetic acid (7-3);
2-(4- { 4- [5-( { [ l-(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- lH-pyrazol-4- yljphenyl }phenyl)-2,2-difluoroacetic acid;
2-(2-fluoro-4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid (9-2);
2-(2-fluoro-4-{4-[l-methyl-5-({ [l-phenylethoxy]carbonyl} amino)- lH-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2-(2-chloro-4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid (9-3);
2- (2-chloro-4- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)acetic acid;
3- (4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)propanoic acid (9-8); and
3-(4-{4-[l-methyl-5-({ [l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)propanoic acid; or
a stereoisomer or pharmaceutically acceptable salt thereof.
[0025] In yet another embodiment, the invention provides a compound selected from the group consisting of:
(R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid (6a);
(R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid potassium salt (6b);
l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid (6a racemic);
l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid potassium salt;
l-(4- { 4- [5-( { [ l-(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l-carboxylic acid (7-1);
l-(4-{2-fluoro-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l-carboxylic acid (7-2);
l-(4-{2-fluoro-4-[l-methyl-5-({ [l-phenylethoxy]carbonyl} amino)- lH-pyrazol-4- yl]phenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{3-methyl-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (7-4);
1 -(4- { 3 -methyl-4- [ 1 -methyl-5 -( { [ 1 -phenylethoxy]carbonyl } amino)- 1 H-pyrazol-4- yl]phenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(2-fluoro-4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l-carboxylic acid (9-1);
1 -(2-fluoro-4- { 4- [ 1 -methyl-5-( { [ 1 -phenylethoxy] carbonyl } amino)- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{3-fluoro-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (9-6);
1 -(4- { 3 -fluoro-4- [ 1 -methyl-5-( { [ 1 -phenylethoxy] carbonyl } amino)- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 -(4- { 2-methyl-4- [ 1 -methyl-5 -( { [( 1R)- 1 -pheny lethoxy]carbonyl } amino)- lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (9-7);
1 -(4- { 2-methyl-4- [ 1 -methyl-5 -( { [ 1 -phenylethoxy] carbonyl } amino)- 1 H-pyrazol-4- yl]phenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{3-methoxy-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol- 4-yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (9-9);
1 -(4- { 3-methoxy-4-[ 1 -methyl-5-({ [ 1 -phenylethoxy]carbonyl } amino)- lH-pyrazol-4- yl]phenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{2-methoxy-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol- 4-yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (9-10);
1 -(4- {2-methoxy-4-[ 1 -methyl-5-({ [ 1 -phenylethoxy]carbonyl } amino)- lH-pyrazol-4- yl]phenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}arnino)-lH-l,2,3-triazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (11);
l-(4-{4-[l-methyl-5-({ [l-phenylethoxy]carbonyl}amino)-lH-l,2,3-triazol-4- yl]phenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{4-[l-methyl-5-({ [(lR)-l-phenylpropoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (12-1);
1 -(4- {4-[ 1 -methyl-5-( { [ l-phenylpropoxy]carbonyl } amino)- lH-pyrazol-4- yl]phenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 - { 4- [4- ( 1 -methyl- 5 - { [( 1 -pheny lbutoxy )carbonyl] amino } - 1 H-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (12-2);
1 -(4- {4-[ 1 -methyl-5-( { [ l-(3-methylphenyl)ethoxy]carbonyl } amino)- lH-pyrazol-4- yl]phenyl} pheny l)cyclopropane-l -carboxylic acid (12-4);
l-(4-{4-[5-({ [(lR)-l-(2-chloro-4-fluorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH- pyrazol-4-yl]phenyl}phenyl)cyclopropane-l-carboxylic acid (12-5);
1 -(4- { 4- [5 -( { [ 1 -(2-chloro-4-fluorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1H- pyrazol-4-yl]phenyl}phenyl)cyclopropane-l-carboxylic acid;
1 - { 4- [2-(hydroxymethyl)-4- [ l-methyl-5-( { [( 1R)- 1 -phenylethoxyjcarbonyl } amino)- 1H- pyrazol-4-yl]phenyl]phenyl } cyclopropane- 1-carboxy lie acid (15);
1 - { 4- [2-(hydroxymethyl)-4- [ l-methyl-5-( { [ 1 -phenylethoxyjcarbonyl } amino)- 1H- pyrazol-4-yl]phenyl]phenyl } cyclopropane- 1-carboxy lie acid;
1 - { 4- [4-( 1 -methyl-5- { [( 1 -phenyl- 1 -deuteroethoxy)carbonyl] amino } - 1 H-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1-carboxy lie acid (18-1);
1 - { 4- [4-( 1 -methyl-5- { [( 1 -phenyl- 1 -deutero- 1 -trideuteromethylmethoxy)carbonyl] amino } - 1 H-pyrazol-4-yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid (18-2);
1 - { 4- [4-( 1 -methyl-5- { [( 1 -pentadeuterophenyl- 1 -deutero- 1 -trideuteromethylmethoxy) carbonyl]amino}-lH-pyrazol-4-yl)phenyl]phenyl}cyclopropane-l -carboxylic acid (18-3);
1 - { 4- [4-(5 - { [(2-methoxy- 1 -phenylethoxy)carbonyl]amino } - 1 -methyl- lH-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (19);
l-{4-[4-(5-{ [(3-methoxy-l-phenylpropoxy)carbonyl]amino}-l-methyl-lH-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (21);
1 - { 4- [4-(5- { [(2-ethoxy- 1 -phenylethoxy)carbonyl] amino } - 1 -methyl- 1 H-pyrazol-4- yl)phenyl]phenyl} cyclopropane- 1 -carboxylic acid (23);
l-(4-{4-[l-methyl-5-({ [l-(2-methylphenyl)ethoxy]carbonyl} amino)- lH-pyrazol-4- yl]phenyl}phenyl)cyclopropane-l -carboxylic acid (26); or
a stereoisomer or pharmaceutically acceptable salt thereof.
[0026] In another embodiment, the invention provides a compound of Formula (1) or a pharmaceutical acceptable salt thereof, wherein the substituents have the meaning given in the Examples. In particular embodiments, the invention provides a potassium salt of a compound of Formula (1).
[0027] In another aspect, the present invention provides a pharmaceutical composition comprising a compound having Formula (1), (2), (3) or (4) or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
[0028] In another aspect, the present invention provides a combination comprising a compound having Formula (1), (2), (3) or (4) or a pharmaceutically acceptable salt thereof, and a
second therapeutic agent.
[0029] In another aspect, the invention provides a method for inhibiting LPA in a cell, comprising contacting the cell with an effective amount of a compound having Formula (1), (2),
(3) or (4) or a pharmaceutically acceptable salt thereof, and optionally in combination with a second therapeutic agent.
[0030] In yet another aspect, the invention provides a method for treating a LPA-dependent or LPA-mediated disease or condition comprising administering to a subject in need thereof a therapeutically effective amount of a compound having Formula (1), (2), (3) or (4) or a pharmaceutically acceptable salt thereof, and optionally in combination with a second therapeutic agent. The invention also provides a compound having Formula (1), (2), (3) or (4) or a pharmaceutically acceptable salt thereof, and optionally in combination with a second therapeutic agent, for use in the treatment of a LPA-dependent or LPA-mediated disease or condition. The invention further provides the use of a compound having Formula (1), (2), (3) or
(4) or a pharmaceutically acceptable salt thereof, and optionally in combination with a second therapeutic agent, for the preparation of a medicament for the treatment of a LPA-dependent or LPA-mediated disease or condition in a subject.
[0031] In any of the aforementioned combination, uses and methods for using the compounds of the invention, the second therapeutic agent can be selected from corticosteroids, immunosuppresant, analgesics, anti-cancer agent, anti-inflammatories, chemokine receptor antagonists, bronchodilators, leukotriene receptor antagonists, leukotriene formation inhibitors, monoacylglycerol kinase inhibitors, phospholipase A] inhibitors, phospholipase A2 inhibitors, and lysophospholipase D (lysoPLD) inhibitors, autotaxin inhibitors, decongestants,
antihistamines, mucolytics, anticholinergics, antitussives, expectorants, and β-2 agonists.
[0032] In any of the aforementioned uses and methods for using the compounds of the invention, the LPA-dependent or LPA-mediated disease or condition can be selected from: (i) pulmonary fibrosis, idiopathic pulmonary fibrosis, iatrogenic drug-induced fibrosis, occupational or environmental induced fibrosis, sarcoidosis, hypersensitivity pneumonia, collagen vascular disease, alveolar proteinosis, langerhans cell granulomatosis, lymphangioleiomyomatosis, Hermansky-Pudlak Syndrome, tuberous sclerosis, neurofibromatosis, metabolic storage disorders and familial interstitial lung disease; (ii) fibrosis of organs or tissues, scarring, liver diseases, dermatological conditions, cancer, cardiovascular diseases, respiratory diseases or conditions, inflammatory diseases, gastrointestinal tract diseases, renal diseases, urinary tract-
associated diseases, inflammatory diseases of lower urinary tract, dysuria, frequent urination, pancreas disease, arterial obstruction, cerebral infarction, cerebral hemorrhage, pain, peripheral neuropathy, and fibromyalgia; (iii) radiation induced fibrosis; chronic obstructive pulmonary disease (COPD), scleroderma, systemic sclerosis, bleomycin induced pulmonary fibrosis, chronic asthma, silicosis, asbestos induced pulmonary fibrosis, acute respiratory distress syndrome (ARDS), kidney fibrosis, tubulointerstitium fibrosis, glomerular nephritis, focal segmental glomerular sclerosis, lupus nephritis, IgA nephropathy, hypertension, Alport, gut fibrosis, liver fibrosis, cirrhosis, alcohol induced liver fibrosis, toxic/drug induced liver fibrosis, hemochromatosis, nonalcoholic steatohepatitis (NASH), biliary duct injury; primary biliary cirrhosis, infection induced liver fibrosis, viral induced liver fibrosis, autoimmune hepatitis, corneal scarring, hypertrophic scarring, Dupuytren's disease, keloids, cutaneous fibrosis, cutaneous scleroderma, spinal cord injury/fibrosis, myelofibrosis, vascular restenosis, atherosclerosis, arteriosclerosis, Wegener's granulomatosis, Peyronie's disease, chronic lymphocytic leukemia, tumor metastasis, transplant organ rejection, endometreosis, neonatal respiratory distress syndrome and neuropathic pain; or (iv) renal fibrosis, acute kidney injury, chronic kidney disease, skin fibrosis, fibrosis of the gut, ocular fibrosis, breast cancer, pancreatic cancer, ovarian cancer, prostate cancer, glioblastoma, bone cancer, colon cancer, bowel cancer, head and neck cancer, melanoma, multiple myeloma, chronic lymphocytic leukemia, cancer pain, tumor metastatis, transplant organ rejection, age related macular degeneration (AMD), diabetic retinopathy, and Raynaud' s phenomenon.
[0033] In specific embodiments, the LPA-dependent or LPA-mediated disease or condition is selected from pulmonary fibrosis, idiopathic pulmonary fibrosis, asthma, chronic obstructive pulmonary disease (COPD), renal fibrosis, acute kidney injury, chronic kidney disease, liver fibrosis, skin fibrosis, fibrosis of the gut, ocular fibrosis, breast cancer, pancreatic cancer, ovarian cancer, prostate cancer, glioblastoma, bone cancer, colon cancer, bowel cancer, head and neck cancer, melanoma, multiple myeloma, chronic lymphocytic leukemia, cancer pain, tumor metastatis, transplant organ rejection, scleroderma, age related macular degeneration (AMD), diabetic retinopathy, collagen vascular disease, atherosclerosis, Raynaud's phenomenon and neuropathic pain; and more particularly, idiopathic pulmonary fibrosis.
Definitions
[0034] For purposes of interpreting this specification, the following definitions will apply and whenever appropriate, terms used in the singular will also include the plural and vice versa.
[0035] As used herein, "Ci-6 alkyl" denotes a an alkyl radical having from 1 up to 6, particularly up to 4 carbon atoms, the radicals being either linear or branched with single or multiple branching; for example, butyl, such as n-butyl, sec -butyl, isobutyl, tert-butyl; propyl, such as n-propyl or isopropyl; ethyl or methyl; more particularly, methyl, propyl or tert-butyl.
[0036] As used herein, "C2^ alkynyl" denotes an alkynyl radical having from 2 up to 4 carbon atoms, the radicals being either linear or branched; for example, ethynyl, prop-l-ynyl, etc.
[0037] As used herein, "Ci_6 alkoxy" refers to Ci_6 alkyl-O-, for example, methoxy, ethoxy, isopropyloxy, or tert-butoxy.
[0038] As used herein, "Ci_6 hydroxyalkyl" refers to Ci_6 alkyl-OH, wherein Ci_6 alkyl is as defined above. The hydroxy group may be attached to the alkyl radical on any carbon within the alkyl radical, for example, hydroxymethyl, 2-hydroxyethyl or 2-hydroxy-2-propyl.
[0039] As used herein, "halogen" or "halo" refers to fluoro, chloro, bromo, and iodo; and more particularly, fluoro or chloro.
[0040] As used herein, "haloCi_6 alkyl" or "Ci_6 haloalkyl" refers to an alkyl radical, as defined above, that is substituted by one or more halo radicals, as defined above, for example, fluoroCi-6 alkyl (e.g., trifluoromethyl).
[0041] As used herein, "halo Ci_6alkoxy" refers to an alkoxy radical, as defined above, that is substituted by one or more halo radicals, as defined above, for example fluoroCi-6 alkoxy (e.g., trifluoromethoxy or difluoromethoxy).
[0042] The term "aryl" as used herein, refers to an aromatic substituent which can be a single aromatic ring, or multiple aromatic rings that are fused together. Non-limiting examples include phenyl, naphthyl or tetrahydronaphthyl.
[0043] As used herein, the term "heterocyclyl" or "heterocyclo" refers to a saturated or unsaturated non-aromatic ring or ring system, e.g. , which is a 4-, 5-, 6-, or 7-membered monocyclic, 7-, 8-, 9-, 10-, 11-, or 12-membered bicyclic or 10-, 11-, 12-, 13-, 14- or 15- membered tricyclic ring system and contains at least one heteroatom selected from O, S and N, where the N and S can also optionally be oxidized to various oxidation states. The heterocyclic group can be attached at a heteroatom or a carbon atom. Examples of heterocycles include tetrahydrofuran (THF), dihydrofuran, 1, 4-dioxane, morpholine, 1,4-dithiane, piperazine, piperidine, 1,3-dioxolane, imidazolidine, imidazoline, pyrroline, pyrrolidine, azetidinyl, tetrahydropyran, dihydropyran, oxathiolane, dithiolane, 1,3-dioxane, 1,3-dithiane, oxathiane,
thiomorpholine, and the like.
[0044] As used herein, the term "heteroaryl" refers to a 5-14 membered monocyclic- or bicyclic- or tricyclic-aromatic ring system, having 1 to 8 heteroatoms. Typically, the heteroaryl is a 5-10 membered ring system (e.g., 5-7 membered monocycle or an 8-10 membered bicycle) or a 5-7 membered ring system. Typical 5-6 membered heteroaryls include 2- or 3-thienyl, 2- or
3- furyl, 2- or 3-pyrrolyl, 2-, 4-, or 5-imidazolyl, 3-, 4-, or 5- pyrazolyl, 2-, 4-, or 5-thiazolyl, 3-,
4- , or 5-isothiazolyl, 2-, 4-, or 5-oxazolyl, 3-, 4-, or 5-isoxazolyl, 3- or 5-1,2,4-triazolyl, 4- or 5- 1,2, 3-triazolyl, tetrazolyl, 2-, 3-, or 4-pyridyl, 3- or 4-pyridazinyl, 3-, 4-, or 5-pyrazinyl, 2- pyrazinyl, and 2-, 4-, or 5-pyrimidinyl.
[0045] As used herein, a "bicyclic" carbocyclic, heterocyclic or heteroaryl ring refers to a carbocyclic, heterocyclic or heteroaryl ring that is fused to one or more aryl, cycloaliphatic or heterocyclyl rings. Fusion of the ring can occur in several ways: i) across a bond between two atoms (e.g., dihydro-indenyl or tetrahydronaphthalenyl); ii) at a single atom forming a spirocyclic compound (e.g., l-oxaspiro[4.5]decanyl); or iii) across a sequence of atoms (i.e., a bridgehead atom), for example, bicycle[2.2.1]heptane. Non-limiting examples of a heteroaromatic ring fused to one or more aryl, cycloaliphatic or heterocyclyl rings, wherein the radical or point of attachment is on the heteroaromatic ring include 1-, 2-, 3-, 5-, 6-, 7-, or 8- indolizinyl, 1-, 3-, 4-, 5-, 6-, or 7-isoindolyl, 2-, 3-, 4-, 5-, 6-, or 7-indolyl, 2-, 3-, 4-, 5-, 6-, or 7-indazolyl, 2-, 4-, 5-, 6-, 7-, or 8- purinyl,
1- , 2-, 3-, 4-, 6-, 7-, 8-, or 9-quinolizinyl, 2-, 3-, 4-, 5-, 6-, 7-, or 8-quinoliyl, 1-, 3-, 4-, 5-, 6-, 7-, or 8- isoquinoliyl, 1-, 4-, 5-, 6-, 7-, or 8-phthalazinyl, 2-, 3-, 4-, 5-, or 6-naphthyridinyl, 2-, 3- , 5-, 6-, 7-, or 8- quinazolinyl, 3-, 4-, 5-, 6-, 7-, or 8-cinnolinyl, 2-, 4-, 6-, or 7-pteridinyl, 1-, 2-, 3-, 4-, 5-, 6-, 7-, or 8-4aH carbazolyl, 1-, 2-, 3-, 4-, 5-, 6-, 7-, or 8-carbzaolyl, 1-, 3-, 4-, 5-, 6-, 7-, 8-, or 9-carbolinyl, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9-, or 10-phenanthridinyl, 1- , 2-, 3-, 4-, 5-, 6-, 7-, 8-, or 9-acridinyl, 1-, 2-, 4-, 5-, 6-, 7-, 8-, or 9-perimidinyl, 2-, 3-, 4-, 5-, 6-, 8-, 9-, or 10-phenathrolinyl, 1-, 2- , 3-, 4-, 6-, 7-, 8-, or 9-phenazinyl, 1-,
2- , 3-, 4-, 6-, 7-, 8-, 9-, or 10-phenothiazinyl, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9-, or 10-phenoxazinyl, 2-, 3-, 4-, 5-,
6- , or 1-, 3-, 4-, 5-, 6-, 7-, 8-, 9-, or 10- benzisoqinolinyl, 2-, 3-, 4-, or thieno[2,3-b]furanyl, 2-, 3-, 5-, 6-,
7- , 8-, 9-, 10 -, or l l-7H-pyrazino[2,3-c]carbazolyl,2-, 3-, 5-, 6-, or 7-2H- furo[3,2-b]-pyranyl, 2-, 3-, 4-,
5- , 7-, or 8-5H-pyrido[2,3-d]-o-oxazinyl, 1-, 3-, or 5-lH-pyrazolo[4,3-d]-oxazolyl, 2-, 4-, or 54H- imidazo[4,5-d] thiazolyl, 3-, 5-, or 8-pyrazino[2,3-d]pyridazinyl, 2-, 3-, 5-, or 6- imidazo[2,l-b] thiazolyl, 1-, 3-, 6-, 7-, 8-, or 9-furo[3,4-c]cinnolinyl, 1-, 2-, 3-, 4-, 5-, 6-, 8-, 9-, 10, or l l-4H-pyrido[2,3- c]carbazolyl, 2-, 3-, 6-, or 7-imidazo[l,2-b] [l,2,4]triazinyl, 7-benzo[b]thienyl, 2-, 4-, 5- , 6-, or 7- benzoxazolyl, 2-, 4-, 5-, 6-, or 7-benzimidazolyl, 2-, 4-, 4-, 5-, 6-, or 7 -benzo thiazolyl, 1-, 2-, 4-, 5-, 6-, 7- , 8-, or 9- benzoxapinyl, 2-, 4-, 5-, 6-, 7-, or 8-benzoxazinyl, 1-, 2-, 3-, 5-, 6-, 7-, 8-, 9-, 10-, or 11-lH- pyrrolo[l,2-b][2]benzazapinyl. Typical fused heteroaryl groups include, but are not limited to 2-, 3-, 4-,
5-, 6-, 7-, or 8-quinolinyl, 1-, 3-, 4-, 5-, 6-, 7-, or 8-isoquinolinyl, 2-, 3-, 4-, 5-, 6-, or 7-indolyl, 2-, 3-, 4-, 5-, 6-, or 7-benzo[b]thienyl, 2-, 4-, 5- , 6-, or 7-benzoxazolyl, 2-, 4-, 5-, 6-, or 7-benzimidazolyl, and 2-, 4-, 5-, 6-, or 7-benzothiazolyl.
[0046] As used herein, a "stereoisomer" refers to a compound made up of the same atoms bonded by the same bonds but having different three-dimensional structures, which are not interchangeable. The present invention contemplates various stereoisomers and mixtures thereof and includes "enantiomers", which refers to two stereoisomers whose molecules are non- superimposeable mirror images of one another.
[0047] As used herein, the term "LPA" refers to lysophosphatidic acid, and generally refers to l-acyl-2-hydroxy-sn-glycero-3 -phosphate. The term "LPA" encompasses saturated, and mono- and poly-unsaturated variants of either the snl or the sn2 regioisomers, and different molecular species of LPA including but not limited to the 16-, 18- and 20- carbon long acyl chains. (Tigyi, Brit. Pharmacol. Soc. 161:241-270 (2010)). The term "LPA" also encompasses other forms of LPA such as 1-alkyl- or 2-acyl-LPA. (Choi et al., Annu. Rev. Pharmacol.
Toxicol. 50: 157-86 (2010)).
[0048] As used herein, the terms "LPA1" (also known as EDG-2), "LPA2", "LPA3", "LPA4", "LPA5" and the like refer to specific G protein-coupled receptors (GPCR) for LPA. LPA binding to its cognate GPCRs (LPA1, LPA2, LP A3, LPA4, LPA5, LPA6) activates intracellular signaling pathways to produce a variety of biological responses.
[0049] As used herein, the term "antagonist" refers to a molecule such as a compound, which diminishes, inhibits, or prevents the action of another molecule or the activity of a receptor site, and includes but is not limited to competitive antagonists, noncompetitive antagonists and uncompetitive antagonists.
[0050] As used herein, the term "LPA-dependent" refers to conditions or disorders that would not occur, or would not occur to the same extent, in the absence of LPA. The term "LPA- mediated", as used herein, refers to refers to conditions or disorders that might occur in the absence of LPA but can occur in the presence of LPA.
[0051] As used herein, the term "pharmaceutically acceptable carrier" includes any and all solvents, dispersion media, coatings, surfactants, antioxidants, preservatives (e.g., antibacterial agents, antifungal agents), isotonic agents, absorption delaying agents, salts, preservatives, drugs, drug stabilizers, binders, excipients, disintegration agents, lubricants, sweetening agents, flavoring agents, dyes, and the like and combinations thereof, as would be known to those
skilled in the art (see, for example, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289- 1329, and subsequent versions thereof). Except insofar as any conventional carrier is incompatible with the active ingredient, its use in the therapeutic or pharmaceutical compositions is contemplated.
[0052] As used herein, the term "therapeutically effective amount" refers to an amount of the compound of Formula (1), (2), (3), (4), (5), (5A), (6) or (7), which is sufficient to achieve the stated effect. Accordingly, a therapeutically effective amount of a compound of Formula (1), (2), (3), (4), (5), (5 A), (6) or (7), used in for the treatment of a condition mediated by LPA will be an amount sufficient for the treatment of the condition mediated by LPA.
[0053] As used herein, the term "subject" refers to an animal or human subject. Typically the animal is a mammal. A subject also refers to for example, primates (e.g., humans, male or female), cows, sheep, goats, horses, dogs, cats, rabbits, rats, mice, fish, birds and the like. In certain embodiments, the subject is a primate. In yet other embodiments, the subject is a human.
[0054] As used herein, the term "treat", "treating" or "treatment" of any disease or disorder refers in one embodiment, to ameliorating the disease or disorder (i.e., slowing or arresting or reducing the development of the disease or at least one of the clinical symptoms thereof). In another embodiment "treat", "treating" or "treatment" refers to alleviating or ameliorating at least one physical parameter including those which may not be discernible by the patient. In yet another embodiment, "treat", "treating" or "treatment" refers to modulating the disease or disorder, either physically, (e.g., stabilization of a discernible symptom), physiologically, (e.g., stabilization of a physical parameter), or both. In yet another embodiment, "treat", "treating" or "treatment" refers to preventing or delaying the onset or development or progression of the disease or disorder.
[0055] As used herein, a subject is "in need of a treatment if such subject would benefit biologically, medically or in quality of life from such treatment.
[0056] As used herein, the term "a," "an," "the" and similar terms used in the context of the present invention (especially in the context of the claims) are to be construed to cover both the singular and plural unless otherwise indicated herein or clearly contradicted by the context.
[0057] The chemical naming protocol and structure diagrams used herein employ and rely on the chemical naming features as utilized by the ChemDraw program (available from
CambridgeSoft Corp., Cambridge, MA). In particular, compound structures and names were derived using Chemdraw Ultra (Version 10.0) and/or ChemAxon Name Generator (JChem
Version 5.3.1.0), or subsequent versions thereof.
Brief Description of the Figures
[0058] Figure 1 depicts the X-ray powder diffraction pattern of (R)-l-(4'-(l-methyl-5-((l- phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid (6a) and (R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid potassium salt (6b).
[0059] Figures 2A and 2B depict the differential scanning calorimetry curve and thermogravimmetric plot respectively, for compound (6b).
Modes of Carrying Out the Invention
[0060] The present invention relates to compositions and methods for modulating the activity of LPA receptors. In one aspect, the present invention relates to compounds which act as inhibitors of LPA, and methods of using such compounds to treat a disease or condition associated with one or more LPA receptors. Various embodiments of the invention are described herein. It will be recognized that features specified in each embodiment may be combined with other specified features to provide further embodiments.
[0061] In one aspect, the invention provides a compound having Formula (1):


Z1 and Z2 are independently CH or N; or Z2 is C if attached to R4a;
R1 is hydrogen, halo, hydroxy or CO2H;
R2 is hydrogen, halo or methyl; or
R1 and R2 together form cyclopropyl, or a monocyclic 4-6 membered heterocycle
comprising 1-2 heteroatoms selected from N, O and S; wherein said 4-6 membered ring is unsubstituted or substituted by 1-2 R9 groups;
R3 is -C02H, -C(0)NR-(CRR')o-2-CN, -C(0)NRS02R10 or -NR-C(0)R10;
R4a and R4b are independently hydrogen, halo, Ci_6 alkyl, Ci_6 alkoxy or Ci_6 hydroxyalkyl;
R5 is methyl or ethyl;
R6 is hydrogen, Ci_6 alkyl, -(CRR')i_2-0(Ci-4 alkyl), C2^ alkynyl or cyclopropyl; R6a is hydrogen or Ci_6 alkyl;
R6b is Ci-6 alkyl; cyclopropyl; phenyl substituted with R7 and R8; or a 5-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O or S;
R7 and R8 are independently hydrogen, Ci^ alkyl or halo;
R9 is -C(0)Rn, -C(0)ORn or S02Rn;
R10 and R11 are independently Ci^ alkyl;
R and R' are independently hydrogen or Ci_6 alkyl;
m and n are independently 1-4; and
p is 0-2; or
a stereoisomer or pharmaceutically acceptable salt thereof.
[0062] In one emb Formula (2):


R1 and R2 together form cyclopropyl;
R3, R4a, R4b, R7, R8, m, n and p are as defined in Formula (1); or
a stereoisomer or pharmaceutically acceptable salt thereof.
[0064] In another embodiment, the invention provides a compound having Formula (1), provided said compound is not a compound of Formula (3).
[0065] In yet another embodiment, the invention provides a compound of Formula (4):
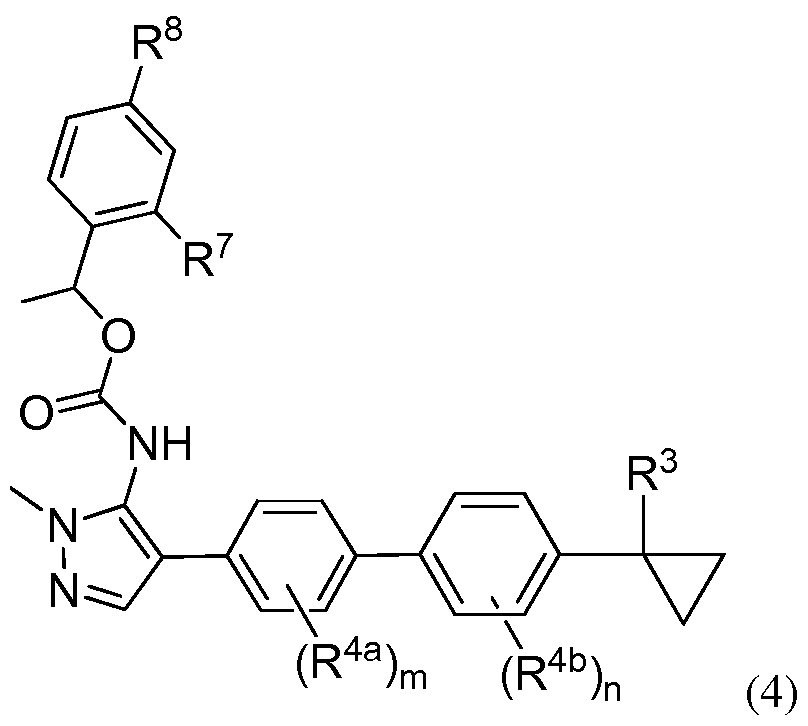
or a stereoisomer or pharmaceutically acceptable salt thereof; wherein the substituents are as defined above.
[0066] In another aspect, the invention provides a compound having Formula (5) or (5 A):
 wherein B is phenyl, C3-7 cycloalkyl, a 4-6 membered heterocycle comprising 1-2 heteroatoms selected from N, NR9, O and S(0)q; N wherein CI of the pyridyl ring is attached to -(CR1R2)P-R3, a 5-6 membered heteroaryl comprising 2-3 heteroatoms selected from N, O and S; or a 9-10 membered bicyclic carbocyclic or bicyclic heterocyclic ring comprising 1-3 heteroatoms selected from N, NR9, O and S;
wherein B is phenyl, C3-7 cycloalkyl, a 4-6 membered heterocycle comprising 1-2 heteroatoms selected from N, NR9, O and S(0)q; N wherein CI of the pyridyl ring is attached to -(CR1R2)P-R3, a 5-6 membered heteroaryl comprising 2-3 heteroatoms selected from N, O and S; or a 9-10 membered bicyclic carbocyclic or bicyclic heterocyclic ring comprising 1-3 heteroatoms selected from N, NR9, O and S;
Z is N;
R1 is hydrogen, Ci_6 alkyl, Ci_3 alkoxy, halo or C02R;
R2 is hydrogen, Ci_6 alkyl or halo; or
R1 and R2 together form cyclopropyl, cyclobutyl, or a 3-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O and S, and wherein said cyclopropyl, cyclobutyl, or 3-6 membered heterocycle is optionally substituted by 1-2 R9;
R3 is -CO2R, cyano, -C(0)NRS02R10, -S02-NR-C(0)R10, -NR-C(0)R10, -NR-SO2R10or tetrazolyl;
R4a and R4b are independently halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONR2 or C02R;
R5 is methyl or ethyl;
R6 is Ci_6 alkyl, Ci_6 haloalkyl or -(CR2)-0(Ci_4 alkyl);
R6a is hydrogen or Ci_6 alkyl;
R6b is Ci_6 alkyl; C3_7 cycloalkyl that is monocyclic or optionally fused to a phenyl that is unsubstituted or substituted with R7 and R8; phenyl substituted with R7 and R8; a 5-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O or S, and unsubstituted or substituted by 1-2 R9; or a 5-6 membered heteroaryl comprising 1-3 heteroatoms selected from N, O and S, and unsubstituted or substituted by 1-2 R9;
R7 and R8 are independently hydrogen, methyl, methoxy or halo;
R9 is (Ci_6 alkyl), -C(0)Rn, -C(0)ORn, -C(0)NRnR12 or S02Rn;
R10, R11 and R12 are independently Ci^ alkyl, haloCi_4 alkyl, cyclopropyl, phenyl unsubstituted or substituted by Ci_4 alkyl;
R is hydrogen or Ci_6 alkyl; and
m, n, p and q are independently 0-2; or
a stereoisomer, prodrug or pharmaceutically acceptable salt thereof.
[0067] In one e ing Formula (6):

[0068] In another embodiment, the invention provides a compound of Formula (5), (5 A) or (6), wherein a substituent is defined independently, collectively, or in any combination or subcombination, as follows:
a) wherein B is phenyl, C3-7 cycloalkyl, a 4-6 membered heterocycle comprising 1-2
heteroatoms selected from N, NR9, O and S(0)q; wherein CI of the pyridyl ring is attached to -(CR1R2)P-R3, a 5-6 membered heteroaryl comprising 2-3 heteroatoms selected from N, O and S; or a 9-10 membered bicyclic carbocyclic or bicyclic heterocyclic ring comprising 1-3 heteroatoms selected from N, NR9, O and S; and particularly, wherein B phenyl, pyrazolyl, cyclohexyl, piperazinyl, 2-oxo- 1 ,2-dihydropyridin-4-yl, cyclopropyl, cyclobutyl, cyclopentyl, azetidinyl, pyrrolidinyl, N wherein CI of the pyridyl ring is attached to -(CR1R2)P-R3, dihydro-indenyl, tetrahydronaphthalenyl, or
tetrahydroisoquinolinyl; wherein the -NH- moiety in said azetidinyl, pyrrolidinyl or tetrahydroisoquinolinyl is optionally substituted by -C(0)Rn or S02Rn and R11 is C1-4 alkyl (e.g., N-acetyl-pyrrolidinyl, N-methylsulfonyl-pyrrolidinyl, N-acetyl-tetrahydroisoquinolinyl and the like); and more particularly, wherein B is phenyl, pyrazolyl, N wherein CI of the pyridyl ring is attached to -(CR1R2)P-R3, cyclohexyl, piperazinyl or 2-oxo- 1,2- dihydropyridin-4-yl ;
b) R1 is hydrogen, Ci_6 alkyl, C1-3 alkoxy, halo or C02R; and particularly, wherein R1 is hydrogen or halo (fluoro);
c) R2 is hydrogen, Ci_6 alkyl or halo; and particularly, wherein R2 is hydrogen or halo (fluoro); or
d) R1 and R2 together form cyclopropyl, cyclobutyl, or a 3-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O and S, and wherein said cyclopropyl, cyclobutyl, or 3-6 membered heterocycle is optionally substituted by 1-2 R9; and particularly, wherein R1 and R2 together form cyclopropyl, oxetanyl, or azetidinyl wherein the hydrogen in NH is optionally replaced by -C(0)Rn or S02Rn and R11 and R12 are independently Ci_4 alkyl (e.g., N-acetyl-azetidinyl or N-methylsulfonyl-azetidinyl);
e) R3 is -C02R, cyano, -C(0)NRS02R10, -S02-NR-C(0)R10, -NR-C(0)R10, -NR-
S02R 10 or tetrazolyl; and particularly, wherein R 3 is-C02R and R is hydrogen or Ci_6 alkyl; and more particularly, wherein R3 is-C02R and R is hydrogen;
f) R4a and R4b if present, are independently halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONR2 or C02R; and particularly, wherein R4a and R4b if present, are
independently halo or Ci_6 alkyl (particularly, fluoro, chloro, or methyl); and more particularly, wherein R4a if present is methyl or fluoro, and R4b if present is fluoro;
g) R5 is methyl or ethyl; and more particularly, wherein R5 is methyl;
h) R6 is Ci_6 alkyl, Ci_6 haloalkyl or -(CR2)-0(C1-4 alkyl); and particularly, wherein R6 is methyl, ethyl, propyl, t-butyl or methoxy methylene;
i) R6a is hydro gen or Ci_6 alkyl (particularly methyl); and particularly, wherein R a is hydrogen;
j) R6b is Ci-6 alkyl; C3-7 cycloalkyl that is monocyclic or optionally fused to a phenyl that is unsubstituted or substituted with R7 and R8 (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, dihydroindenyl); phenyl substituted with R7 and R8; a 5-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O or S, and unsubstituted or substituted by 1-2 R9 (e.g., tetrahydrofuranyl, tetrahydropyranyl, piperidyl, N-Boc piperidyl); or a 5-6 membered heteroaryl comprising 1-3 heteroatoms selected from N, O and S, and unsubstituted or substituted by 1-2 R9 (e.g., pyridyl, oxazolyl, pyrazolyl, furanyl, thiazolyl); and particularly, wherein R6b is isopropyl, t-butyl, methyl, cyclopropyl or phenyl substituted with R7 and R8; k) R7 and R8 are independently hydrogen, methyl, methoxy or halo; and particularly, wherein R7 and R8 are independently hydrogen or halo; and more particularly, wherein R7 is hydrogen and chloro, and R8 is hydrogen or fluoro;
1) R9 is (Ci_6 alkyl), -C(0)Rn, -C(0)ORn, -C(0)NRnR12 or S02Rn;
m) R10, R11 and R12 are independently Ci^ alkyl, haloCi^ alkyl, cyclopropyl, phenyl unsubstituted or substituted by Ci_4 alkyl;
n) R is hydrogen or Ci_6 alkyl;
o) m and n are 0-2; and more particularly, wherein m and n are 0-1 ; and
p) p and q are independently 0-2; or
a stereoisomer, prodrug or pharmaceutically acceptable salt thereof.
[0069] In another embodiment, the invention provides a compound of Formula (7),

wherein m and n are independently 0- 1 ; and
R3, R4a, R4b, R5, R6, R7 and R8 are as defined in Formula (1); or
a stereoisomer, prodrug or pharmaceutically acceptable salt thereof.
[0070] In another embodiment, the invention provides a compound of Formula (7),
wherein m and n are independently 0- 1 ; and
R3, R4a, R4b, R5, R6, R7 and R8 are as defined independently, collectively, or in any combination or sub-combination as follows:
a) R3 is -C02R, cyano, -C(0)NRS02R10, -S02-NR-C(0)R10, -NR-C(0)R10, -NR- SO2R10 or tetrazolyl; and particularly, wherein R3 is-C02R and R is hydrogen or Ci_6 alkyl; and more particularly, wherein R3 is-C02R and R is hydrogen;
b) R4a and R4b if present, are independently halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONR2 or C02R; and particularly, wherein R4a and R4b if present, are independently halo or Ci_6 alkyl (particularly, fluoro, chloro, or methyl); and more particularly, wherein R4a if present is methyl or fluoro, and R4b if present is fluoro;
c) R5 is methyl or ethyl; and more particularly, wherein R5 is methyl;
d) R6 is Ci-6 alkyl, Ci_6 haloalkyl or
 alkyl ); and particularly, wherein R6 is methyl, ethyl, propyl, t-butyl or methoxymethylene;
alkyl ); and particularly, wherein R6 is methyl, ethyl, propyl, t-butyl or methoxymethylene;
e) R7 and R8 are independently hydrogen, methyl, methoxy or halo; and particularly, wherein R7 and R8 are independently hydrogen, methyl or halo; and more particularly, wherein R7 is hydrogen and chloro, and R8 is hydrogen or fluoro;
f) R10 is Ci-4 alkyl, haloCi^ alkyl, cyclopropyl, phenyl unsubstituted or substituted by alkyl;
g) R is hydrogen or Ci_6 alkyl; or
a stereoisomer, prodrug or pharmaceutically acceptable salt thereof.
[0071] In another embodiment, the invention provides a compound of Formula (5A), wherein Z is N; and B, R1, R2, R3, R4a, R4b, R5, R6, R6a, R6b, m and n are as defined
independently, collectively, or in any combination or sub-combination as defined in Formula (5) or (6) above. In particular embodiments, the invention provides a compound of Formula (5 A), wherein:
Z is N;
B is phenyl;
R1 and R2 together form cyclopropyl;
R3 is -C02R;
R6b is phenyl substituted with R7 and R8;
R7 and R8 are independently hydrogen or halo;
R is H; and
R4a, R4b, R5, R6, R6a, m and n are as defined in Formula (1).
[0072] In another embodiment, the invention provides a compound of Formula (5), (5 A) or (6), wherein R1 and R2 are independently hydrogen or fluoro.
[0073] In one further embodiment, the invention provides a compound having Formula (5'), (5B) or (5C):


B is phenyl, C3-7 cycloalkyl, a 4-7 membered heterocycle comprising 1-2 heteroatoms selected from N, NR9, O and S(0)q; a 5-6 membered heteroaryl comprising 1-3 heteroatoms selected from N, O and S; or a 9-12 membered bicyclic carbocyclic or bicyclic heterocyclic ring comprising 1-3 heteroatoms selected from N, NR9, O and S;
L1 and L2 are independently a bond or (CR2);
R1 is hydrogen, Ci_6 alkyl, Ci_3 alkoxy, halo or C02R;
R2 is hydrogen, Ci_6 alkyl or halo; or
R1 and R2 together form cyclopropyl, cyclobutyl, or a 3-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O and S, and wherein said cyclopropyl, cyclobutyl, or 3-6 membered heterocycle is optionally substituted by 1-2 R9;
R3 is -C02R, cyano, -C(0)NRS02R10, S02NR2, -S02-NR-C(0)R10, -NR-C(0)R10, -NR- SO2R10or tetrazolyl;
R4a and R4b are independently halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONR2 or C02R;
R5 is methyl or ethyl;
R6 is Ci_6 alkyl, Ci_6 haloalkyl or -(CR2)-0(Ci_4 alkyl);
R6a is hydrogen or Ci_6 alkyl;
R6b is Ci-6 alkyl; C3-7 cycloalkyl that is monocyclic or optionally fused to a phenyl that is unsubstituted or substituted with R7 and R8; phenyl substituted with R7 and R8; a 5-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O or S, and unsubstituted or substituted by 1-2 R9; or a 5-6 membered heteroaryl comprising 1-3 heteroatoms selected from N, O and S, and unsubstituted or substituted by 1-2 R9;
R7 and R8 are independently hydrogen, methyl, methoxy or halo;
R9 is (Ci_6 alkyl), -C(0)Rn, -C(0)ORn, -C(0)NRnR12 or S02Rn;
R10, R11 and R12 are independently Ci^ alkyl, haloCi-4 alkyl, cyclopropyl, phenyl unsubstituted or substituted by Ci_4 alkyl;
R is hydrogen or Ci_6 alkyl; and
k is 0-1;
m, n, p and q are independently 0-2; or
a stereoisomer, prodrug or pharmaceutically acceptable salt thereof.
[0074] In a second further embodiment, the invention provides a compound of Formula (5'), (5B) or (5C), wherein a substituent is defined independently, collectively, or in any combination or sub-combination, as follows:
a) wherein B is phenyl, C3-7 cycloalkyl, a 4-6 membered heterocycle comprising 1-2 heteroatoms selected from N, NR9, O and S(0)q; wherein CI of the pyridyl ring is attached to
 a 5-6 membered heteroaryl comprising 2-3 heteroatoms selected from N, O and S; or a 9-10 membered bicyclic carbocyclic or bicyclic heterocyclic ring comprising 1-3 heteroatoms selected from N, NR9, O and S; and particularly, wherein B is phenyl, pyrazolyl, cyclohexyl, piperazinyl, 2-oxo-l,2-dihydropyridin-
a 5-6 membered heteroaryl comprising 2-3 heteroatoms selected from N, O and S; or a 9-10 membered bicyclic carbocyclic or bicyclic heterocyclic ring comprising 1-3 heteroatoms selected from N, NR9, O and S; and particularly, wherein B is phenyl, pyrazolyl, cyclohexyl, piperazinyl, 2-oxo-l,2-dihydropyridin-
4-yl, cyclopropyl, cyclobutyl, cyclopentyl, azetidinyl, pyrrolidinyl, wherein CI of the pyridyl ring is attached to -(CR1R2)P-R3, -(CR1R2)k-L1-R3 or -L2-(CR1R2)k-R3, dihydro- indenyl, tetrahydronaphthalenyl, or tetrahydroisoquinolinyl; wherein the -NH- moiety in said azetidinyl, pyrrolidinyl or tetrahydroisoquinolinyl is optionally substituted by -C(0)Rn or S02Rn and R11 is C1-4 alkyl (e.g., N-acetyl-pyrrolidinyl, N-methylsulfonyl-pyrrolidinyl, N- acetyl-tetr like); and more particularly, wherein B is phenyl, pyrazolyl,
 of the pyridyl ring is attached to -(CR1R2)P-R3, -
of the pyridyl ring is attached to -(CR1R2)P-R3, -
 cyclohexyl, piperazinyl or 2-oxo-l,2-dihydropyridin-4-yl; b) R1 is hydrogen, Ci_6 alkyl, C1-3 alkoxy, halo or C02R; and particularly, wherein R1 is hydrogen or halo (fluoro);
cyclohexyl, piperazinyl or 2-oxo-l,2-dihydropyridin-4-yl; b) R1 is hydrogen, Ci_6 alkyl, C1-3 alkoxy, halo or C02R; and particularly, wherein R1 is hydrogen or halo (fluoro);
c) R2 is hydrogen, Ci_6 alkyl or halo; and particularly, wherein R2 is hydrogen or halo (fluoro); or
d) R1 and R2 together form cyclopropyl, cyclobutyl, or a 3-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O and S, and wherein said cyclopropyl, cyclobutyl, or 3-6 membered heterocycle is optionally substituted by 1-2 R9; and particularly, wherein R1 and R2 together form cyclopropyl, oxetanyl, or azetidinyl wherein the hydrogen in NH is optionally replaced by -C(0)Rn or S02Rn and R11 and R12 are independently Ci_4 alkyl (e.g., N-acetyl-azetidinyl or N-methylsulfonyl-azetidinyl);
e) R3 is -C02R, cyano, -C(0)NRS02R10, -S02-NR-C(0)R10, -NR-C(0)R10, -NR- SO2R10or tetrazolyl; and particularly, wherein R3 is-C02R and R is hydrogen or Ci_6 alkyl; and more particularly, wherein R3 is-C02R and R is hydrogen;
f) R4a and R4b if present, are independently halogen, Ci_6 alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONR2 or C02R; and particularly, wherein R4a and R4b if present, are independently halo, Ci_6 alkyl or haloCi_6 alkyl (particularly, fluoro, chloro, methyl or trifluoromethyl); and more particularly, wherein R4a if present is methyl or fluoro, and R4b if present is fluoro;
g) R5 is methyl or ethyl; and more particularly, wherein R5 is methyl;
h) R6 is Ci-6 alkyl, Ci_6 haloalkyl or
 alkyl); and particularly, wherein R6 is methyl, ethyl, propyl, t-butyl or methoxy methylene;
alkyl); and particularly, wherein R6 is methyl, ethyl, propyl, t-butyl or methoxy methylene;
i) R6a is hydro gen or Ci_6 alkyl (particularly methyl); and particularly, wherein R a is hydrogen;
j) R6b is Ci-6 alkyl; C3-7 cycloalkyl that is monocyclic or optionally fused to a phenyl that is unsubstituted or substituted with R7 and R8 (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, dihydroindenyl); phenyl substituted with R7 and R8; a 5-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O or S, and unsubstituted or substituted by 1-2 R9 (e.g., tetrahydrofuranyl, tetrahydropyranyl, piperidyl, N-Boc piperidyl); or a 5-6 membered heteroaryl comprising 1-3 heteroatoms selected from N, O and S, and unsubstituted or substituted by 1-2 R9 (e.g., pyridyl, oxazolyl, pyrazolyl, furanyl, thiazolyl); and particularly, wherein R6b is isopropyl, t-butyl, methyl, cyclopropyl or phenyl substituted with R7 and R8; k) R7 and R8 are independently hydrogen, methyl, methoxy or halo; and particularly, wherein R7 and R8 are independently hydrogen or halo; and more particularly, wherein R7 is hydrogen and chloro, and R8 is hydrogen or fluoro;
1) R9 is (Ci_6 alkyl), -C(0)Rn, -C(0)ORn, -C(0)NRnR12 or S02Rn;
m) R10, R11 and R12 are independently Ci^ alkyl, haloCi^ alkyl, cyclopropyl, phenyl
unsubstituted or substituted by C1-4 alkyl;
n) R is hydrogen or Ci_6 alkyl;
o) m and n are 0-2; and more particularly, wherein m and n are 0-1 ; and
p) p and q are independently 0-2.
[0075] In a third further embodiment, the invention provides a compound having Formula (8):

R1 is Ci-6 alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, hydroxy, hydroxyCi-6 alkyl, halogen, cyano, acetonitrileoxy, NR2, C02R, OS02R10, S02NR-L1-R9,-L-NRC(0)R10, -L- NRS02R9, -L-NRC02R10, L-NRSOz-I^-COzR or -NRS02NR2; or R1 is aryl, C5_7 cycloalkyl, or 4-6 membered heterocyclyl containing 1-3 heteroatoms selected from N, O and S, , each of which is optionally substituted with -L-C02R or -L-tetrazolyl;
R2 is hydrogen, halogen or Ci_6 alkoxy; or
R1 and R2 together with Ring A form a bicyclic 9-10 membered aryl or heteroaryl, said heteroaryl containing 1-3 heteroatoms selected from N, O and S;
R3 is halogen, Ci_6 alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONRR9 or
C02R9;
R4 is hydrogen, cyclopropyl-Ci_2 alkyl or Ci_6 alkyl optionally substituted with 1-3 groups selected from cyano, halogen, hydroxy, Ci_6 alkoxy, C02R or NR2;
R5 and R6 are independently hydrogen, Ci_6 alkyl or haloCi_6 alkyl; or R5 and R6 together with the carbon atoms to which they are attached form a C3-7 cycloalkyl or 5-6 membered heterocyclyl containing 1-2 heteroatoms selected from N, O and S;
R7 is phenyl, 5-6 membered heteroaryl containing 1-3 heteroatoms selected from N, O and S; 5-6 membered heterocyclyl containing 1-2 heteroatoms selected from N, O and S; or a
bicyclic 9-10 membered aryl or heteroaryl, said heteroaryl containing 1-3 heteroatoms selected from N, O and S; wherein R7 is optionally substituted with R11 and 1-2 R12;
R8 is hydrogen, halogen, cyano, Ci_6alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, C(0)NRR10 or C02R10;
R9 is Ci-6 alkyl, haloCi_6 alkyl, C3-7 cycloalkyl or phenyl; wherein said C3-7 cycloalkyl, phenyl is optionally substituted with Ci_6 alkyl, C02R or tetrazolyl;
R10 is Ci_6 alkyl or haloCi_6 alkyl;
R11 is halogen or Ci_6 alkyl;
R12 is halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkyl, haloCi_6 alkoxy or cyano;
R is H or Ci-6 alkyl;
alternatively, R and R9 together with the nitrogen atom in S02NRR9, or R and R10 together with the nitrogen atom in C(0)NRR10 form a 5-6 membered heterocyclic ring;
L and L1 are independently a bond or (CRaRa)k;
Ra is H, Ci-6 alkyl, hydroxy, halo or C1-3 alkoxy;
k is 0-4;
m is 0-2; or
a stereoisomer or pharmaceutically acceptable salt thereof.
[0076] In a fourth further embodiment, the invention provides a compound having Formula
(9 A), (9B), (9

[0077] In a fifth further embodiment, the invention provides a compound having Formula (10

R12 is halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkyl, haloCi_6 alkoxy or cyano;
n is 0-2; and
R1, R2, R3, R4, R5, R6 and R8 are as defined in Formula (8); or
a stereoisomer or pharmaceutically acceptable salt thereof.
[0078] In a sixth further embodiment, the invention provides a compound of Formula (8), (9A), (9B), (9C), (9D), (10A) or (10B), wherein a substituent is defined independently, collectively, or in any combination or sub-combination, as follows:
a) A is phenyl or 5-6 membered heteroaryl containing 1-3 heteroatoms selected from N, O and S; and particularly, wherein A is phenyl, pyridyl or thiazolyl;
b) R1 is Ci-6 alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, hydroxy, hydroxyCi-6 alkyl, halogen, cyano, acetonitrileoxy, NR2, C02R, OS02R10, S02NR-L1-R9,-L-NRC(0)R10, -L- NRS02R9, -L-NRC02R10, L-NRS02-L1-C02R or -NRS02NR2; or R1 is aryl, C5-7 cycloalkyl, or 4-6 membered heterocyclyl containing 1-3 heteroatoms selected from N, O and S, , each of which is optionally substituted with -L-C02R or -L-tetrazolyl; and particularly, wherein R1 is Ci-6 alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, hydroxy, hydroxyCi-6 alkyl, halogen, cyano, acetonitrileoxy, amino, methylamino, C02R, OS02R10, S02NRR9,-L-NRC(0)R10, -L- NRS02R9, -L-NRCO2R10 or L-NRSOz-I^-COzR; or R1 is phenyl, C4_6 cycloalkyl, piperazinyl, 2- oxo- 1,2-dihydropyridinyl, piperidyl, pyrrolidinyl or azetidinyl, each of which is optionally substituted with cyano or -L-C02R; wherein
R9 is Ci-6 alkyl, haloCi_6 alkyl, cyclopropyl, phenyl optionally substituted with Ci_6 alkyl;
R is Ci-6 alkyl or haloCi-6 alkyl;
L and L1 are independently a bond or (CRaRa)k and k is 0-2;
R and Ra are independently H or Ci_6 alkyl;
c) R2 is hydrogen, halogen or Ci_6 alkoxy; and particularly, wherein R2 is hydrogen, methoxy or fluoro;
d) alternatively, R1 and R2 together with Ring A form a bicyclic 9-10 membered aryl or heteroaryl, said heteroaryl containing 1-3 heteroatoms selected from N, O and S; and particularly, wherein R1 and R2 together with Ring A form an indole, indazole, benzimidazole or benzotriazolyl;
e) R3 is halogen, Ci_6 alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, cyano, CONRR9 or C02R9 and m is 0-2; and particularly, wherein m is 0;
f) R4 is hydrogen, cyclopropyl-Ci-2 alkyl or Ci_6 alkyl optionally substituted with 1-3 groups selected from cyano, halogen, hydroxy, Ci_6 alkoxy, C02R or NR2; and particularly, wherein R4 is methyl, ethyl, isopropyl, propyl, or cyanomethyl;
g) R5 and R6 are independently hydrogen, Ci_6 alkyl or haloCi_6 alkyl; or R5 and R6 together with the carbon atoms to which they are attached form a C3-7 cycloalkyl or 5-6 membered heterocyclyl containing 1-2 heteroatoms selected from N, O and S; and particularly, wherein R5 is hydrogen and R6 is hydrogen or methyl;
h) R7 is phenyl, 5-6 membered heteroaryl containing 1-3 heteroatoms selected from N, O and S; 5-6 membered heterocyclyl containing 1-2 heteroatoms selected from N, O and S; or a bicyclic 9-10 membered aryl or heteroaryl, said heteroaryl containing 1-3 heteroatoms selected from N, O and S; wherein R7 is optionally substituted with R11 and 1-2 R12; and particularly, wherein R7 is phenyl optionally substituted with R11 and 1-2 R12; and wherein R12 is halogen or Ci_6 alkyl;
i) R8 is hydrogen, halogen, cyano, Ci_6alkyl, haloCi_6 alkyl, Ci_6 alkoxy, haloCi_6 alkoxy, C(0)NRR10 or C02R10; and particularly, wherein R8 is hydrogen;
j) R9 is Ci-6 alkyl, haloCi_6 alkyl, C3-7 cycloalkyl or phenyl; wherein said C3-7 cycloalkyl, phenyl is optionally substituted with Ci_6 alkyl, C02R or tetrazolyl; and particularly, wherein R9 is Ci_6 alkyl, haloCi_6 alkyl, cyclopropyl, phenyl optionally substituted with Ci_6 alkyl;
k) R10 is Ci_6 alkyl or haloCi_6 alkyl;
1) R11 is halo gen or Ci_6 alkyl; and particularly, wherein R11 is chloro or methyl;
m) R is halogen, Ci_6 alkyl, Ci_6 alkoxy, haloCi_6 alkyl, haloCi_6 alkoxy or cyano; and particularly, wherein R12 is fluoro;
n) R is H or Ci_6 alkyl; and
o) Ra is H, Ci_6 alkyl, hydroxy, halo or Ci_3 alkoxy; and particularly, wherein Ra is H or d_6 alkyl.
[0079] In a seventh further embodiment, the invention provides a compound of Formula (8), (9A), (9B), (9C), (9D), (10A) or (10B), wherein R2 is hydrogen, methoxy or fluoro; and m is 0. In other embodiments, R5 is hydrogen; R6 is hydrogen or methyl; and R8 is hydrogen. In yet other embodiments, R11 is chloro or methyl; n is 0-1 ; and R12 is fluoro.
[0080] Unless specified otherwise, the term "compound(s) of the present invention" refers to a compound of Formula (1), (2), (3), (4), (5), (5 A), (5'), (5B), (5C), (6), (7), (8), (9 A), (9B), (9C), (9D), (10A), (10B) or any combination thereof, prodrugs thereof, salts of the compound and/or prodrugs, hydrates or solvates of the compounds, salts and/or prodrugs, as well as all stereoisomers (including diastereoisomers and enantiomers), tautomers and isotopically labeled compounds (including deuterium substitutions), as well as inherently formed moieties (e.g., polymorphs, solvates and/or hydrates).
[0081] Certain of the compounds described herein contain one or more asymmetric centers or axes and may thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)-. The present invention is meant to include all possible isomers, including racemic mixtures, optically pure forms and intermediate mixtures. Optically active (R)- and (S)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques. If the compound contains a double bond, the substituent may be E or Z configuration. If the compound contains a disubstituted cycloalkyl, the cycloalkyl substituent may have a cis- or trans-configuration. All tautomeric forms are also intended to be included.
[0082] Any formula given herein is also intended to represent unlabeled forms as well as isotopically labeled forms of the compounds. Isotopically labeled compounds have structures depicted by the formulas given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number. Examples of isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as 2H, 3H, nC, 13C, 14C, 15N, 18F, 31P, 32P, 35S, 36C1 and
125
I respectively. The invention includes various isotopically labeled compounds as defined
herein, for example those into which radioactive isotopes, such as 3H, 13C, and 14C , are present. Such isotopically labelled compounds are useful in metabolic studies (with 14C), reaction kinetic studies (with, for example 2H or 3H), detection or imaging techniques, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT) including drug or
18 substrate tissue distribution assays, or in radioactive treatment of patients. In particular, an F or labeled compound may be particularly desirable for PET or SPECT studies. Isotopically labeled compounds of this invention and prodrugs thereof can generally be prepared by carrying out the procedures disclosed in the schemes or in the examples and preparations described below by substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent.
[0083] Further, substitution with heavier isotopes, particularly deuterium (i.e., 2H or D) may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements or an improvement in therapeutic index. It is understood that deuterium in this context is regarded as a substituent of a compound of the formula (I). The concentration of such a heavier isotope, specifically deuterium, may be defined by the isotopic enrichment factor. The term "isotopic enrichment factor" as used herein means the ratio between the isotopic abundance and the natural abundance of a specified isotope. If a substituent in a compound of this invention is denoted deuterium, such compound has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium incorporation at each designated deuterium atom), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium incorporation), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5% deuterium incorporation).
[0084] Isotopically-labeled compounds of the invention can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Processes using an appropriate isotopically- labeled reagents in place of the non-labeled reagent previously employed.
[0085] Pharmaceutically acceptable solvates in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D20, d6-acetone, d6- DMSO.
[0086] Compounds of the invention that contain groups capable of acting as donors and/or acceptors for hydrogen bonds may be capable of forming co-crystals with suitable co-crystal formers. These co-crystals may be prepared from the compounds of the invention by known co- crystal forming procedures. Such procedures include grinding, heating, co-subliming, co- melting, or contacting in solution the compounds of the invention with the co-crystal former under crystallization conditions and isolating co-crystals thereby formed. Suitable co-crystal formers include those described in WO 2004/078163. Hence the invention further provides co- crystals comprising a compound of the invention.
[0087] Any asymmetric atom (e.g., carbon or the like) of the compound(s) of the present invention can be present in racemic or enantiomerically enriched, for example the (R)-, (S)- or (R,S)- configuration. In certain embodiments, each asymmetric atom has at least 50 % enantiomeric excess, at least 60 % enantiomeric excess, at least 70 % enantiomeric excess, at least 80 % enantiomeric excess, at least 90 % enantiomeric excess, at least 95 % enantiomeric excess, or at least 99 % enantiomeric excess in the (R)- or (S)- configuration. Substituents at atoms with unsaturated bonds may, if possible, be present in cis- (Z)- or trans- (E)- form.
[0088] Accordingly, as used herein a compound of the present invention can be in the form of one of the possible isomers, rotamers, atropisomers, tautomers or mixtures thereof, for example, as substantially pure geometric (cis or trans) isomers, diastereomers, optical isomers (antipodes), racemates or mixtures thereof. Any resulting mixtures of isomers can be separated on the basis of the physicochemical differences of the constituents, into the pure or substantially pure geometric or optical isomers, diastereomers, racemates, for example, by chromatography and/or fractional crystallization. Any resulting racemates of final products or intermediates can be resolved into the optical antipodes by known methods, e.g., by separation of the
diastereomeric salts thereof, obtained with an optically active acid or base, and liberating the optically active acidic or basic compound. In particular, a basic moiety may thus be employed to resolve the compounds of the present invention into their optical antipodes, e.g., by fractional crystallization of a salt formed with an optically active acid, e.g., tartaric acid, dibenzoyl tartaric acid, diacetyl tartaric acid, di-0,0'-p-toluoyl tartaric acid, mandelic acid, malic acid or camphor- 10-sulfonic acid. Racemic products can also be resolved by chiral chromatography, e.g., high pressure liquid chromatography (HPLC) using a chiral adsorbent.
Pharmacology and Utility
[0089] The compounds of the invention in free form or in salt form, exhibit valuable pharmacological properties, e.g. LPA modulating properties, e.g. as indicated in in vitro and/or in vivo tests as provided in the next sections, and are therefore indicated for therapy in treating a disorder which may be treated by modulating LPA, such as those described below.
[0090] In one aspect, presented herein are compounds of Formula (1), (2), (3), (4), (5), (5A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B), that inhibit the
physiological activity of lysophosphatidic acid (LPA), and therefore, are useful as agents for the treatment or prevention of diseases in which inhibition of the physiological activity of LPA is useful, such as diseases in which an LPA receptor participates, is involved in the etiology or pathology of the disease, or is otherwise associated with at least one symptom of the disease. In one embodiment, the compounds of Formula (1), (2), (3), (4), (5), (5 A), (5'), (5B), (5C), (6), (7), (8), (9 A), (9B), (9C), (9D), (10A) or (10B), are antagonists of at least one of the LPA receptors selected from LPAl, LPA2, LPA3, LPA4, LPA5 and LPA6, and more particularly antagonists of LPAl.
[0091] The compounds of the invention or a pharmaceutically acceptable salt thereof are useful in the treatment of diseases, disorders, or conditions in which activation of at least one LPA receptor by LPA contributes to the symptomology or progression of the disease, disorder or condition. These diseases, disorders, or conditions may arise from one or more of a genetic, iatrogenic, immunological, infectious, metabolic, oncological, toxic, surgical, and/or traumatic etiology. In one aspect, the methods, compounds, pharmaceutical compositions, and
medicaments described herein comprise antagonists of LPA receptors. In one aspect, the methods, compounds, pharmaceutical compositions, and medicaments described herein comprise antagonists of LPAl.
[0092] In one embodiment, the compounds of the invention are useful for the treatment of a LPA-dependent or LPA-mediated disease or condition selected from pulmonary fibrosis, idiopathic pulmonary fibrosis, a diffuse parenchymal interstitial lung disease including iatrogenic drug-induced fibrosis, occupational and/or environmental induced fibrosis (Farmer lung), granulomatous diseases (sarcoidosis, hypersensitivity pneumonia), collagen vascular disease (scleroderma), alveolar proteinosis, langerhans cell granulomatosis,
lymphangioleiomyomatosis, Hermansky-Pudlak Syndrome, tuberous sclerosis,
neurofibromatosis, metabolic storage disorders, or familial interstitial lung disease.
[0093] In another embodiment, the compounds of the invention or a pharmaceutically acceptable salt thereof are useful for the treatment of a LPA-dependent or LPA-mediated disease or condition selected from fibrosis of organs or tissues, scarring, liver diseases, dermatological conditions, cancer, cardiovascular disease, respiratory diseases or conditions including asthma, chronic obstructive pulmonary disease (COPD), pulmonary fibrosis, pulmonary arterial hypertension or acute respiratory distress syndrome; inflammatory disease, gastrointestinal tract disease, renal disease, urinary tract-associated disease, inflammatory disease of lower urinary tract, dysuria, frequent urination, pancreas disease, arterial obstruction, cerebral infarction, cerebral hemorrhage, pain, peripheral neuropathy, and fibromyalgia.
[0094] In other embodiments, the compounds of the invention are useful for the treatment of a LPA-dependent or LPA-mediated disease or condition selected from radiation induced fibrosis; chronic obstructive pulmonary disease (COPD), scleroderma, systemic sclerosis, bleomycin induced pulmonary fibrosis, chronic asthma, silicosis, asbestos induced pulmonary fibrosis, acute respiratory distress syndrome (ARDS), kidney fibrosis, tubulointerstitium fibrosis, glomerular nephritis, focal segmental glomerular sclerosis, lupus nephritis, IgA nephropathy, hypertension, Alport, gut fibrosis, liver fibrosis, cirrhosis, alcohol induced liver fibrosis, toxic/drug induced liver fibrosis, hemochromatosis, nonalcoholic steatohepatitis (NASH), biliary duct injury; primary biliary cirrhosis, infection induced liver fibrosis, viral induced liver fibrosis, autoimmune hepatitis, corneal scarring, hypertrophic scarring, Dupuytren's disease, keloids, cutaneous fibrosis, cutaneous scleroderma, spinal cord injury/fibrosis, myelofibrosis, vascular restenosis, atherosclerosis, arteriosclerosis, Wegener's granulomatosis, Peyronie's disease, chronic lymphocytic leukemia, tumor metastasis, transplant organ rejection, endometreosis, neonatal respiratory distress syndrome, neuropathic pain.or other LPA-dependent or LPA- mediated disease (see e.g., Tigyi, British J Pharmacology vol 161, 241-270, 2010).
[0095] In another embodiment, the compounds of the invention or a pharmaceutically acceptable salt thereof are useful for the treatment of a LPA-dependent or LPA-mediated disease or condition selected from fibrosis of organs (liver, kidney, lung, heart and the like), liver diseases (acute hepatitis, chronic hepatitis, liver fibrosis, liver cirrhosis, portal hypertension, regenerative failure, liver hypofunction, hepatic blood flow disorder, and the like), cell proliferative disease (cancer (solid tumor, solid tumor metastasis, vascular fibroma, myeloma, multiple myeloma, Kaposi's sarcoma, leukemia, chronic lymphocytic leukemia (CLL and the like) and invasive metastasis of cancer cell, and the like), inflammatory disease (psoriasis,
nephropathy, pneumonia and the like), gastrointestinal tract disease (irritable bowel syndrome (IBS), inflammatory bowel disease (IBD), abnormal pancreatic secretion, and the like), renal disease, urinary tract-associated disease (benign prostatic hyperplasia or symptoms associated with neuropathic bladder disease, spinal cord tumor, hernia of intervertebral disk, spinal canal stenosis, symptoms derived from diabetes, lower urinary tract disease (obstruction of lower urinary tract, and the like), inflammatory disease of lower urinary tract, dysuria, frequent urination, and the like), pancreas disease, abnormal angiogenesis- associated disease (arterial obstruction and the like), scleroderma, brain-associated disease (cerebral infarction, cerebral hemorrhage, and the like), neuropathic pain, peripheral neuropathy, and the like, ocular disease (age-related macular degeneration (AMD), diabetic retinopathy, proliferative vitreoretinopathy (PVR), cicatricial pemphigoid, glaucoma filtration surgery scarring, and the like).
[0096] In yet other embodiments, the compounds of the invention or a pharmaceutically acceptable salt thereof are useful for the treatment of a LPA-dependent or LPA-mediated disease or condition selected from renal fibrosis (tubulointerstitium fibrosis, glomerular sclerosis), acute kidney injury, chronic kidney disease, skin fibrosis, fibrosis of the gut, ocular fibrosis, breast cancer, pancreatic cancer, ovarian cancer, prostate cancer, glioblastoma, bone cancer, colon cancer, bowel cancer, head and neck cancer, melanoma, multiple myeloma, chronic lymphocytic leukemia, cancer pain, tumor metastatis, transplant organ rejection, age related macular degeneration (AMD), diabetic retinopathy, and Raynaud's phenomenon. Compounds disclosed herein are also useful for the treatment of post-transplant fibrosis associated with chronic rejection (bronchiolitis obliterans for lung transplant), cutaneous fibrosis (cutaneous scleroderma, Dupuytren disease, keloids), hepatic fibrosis with or without cirrhosis (toxic/drug induced hemochromatosis), alcoholic liver disease, viral hepatitis (hepatitis B virus, hepatitis C virus, HCV), metabolic and auto-immune diseases.
[0097] In one embodiment, the invention provides a method for the treatment or prevention of pulmonary fibrosis (idiopathic pulmonary fibrosis), renal fibrosis, hepatic fibrosis, ocular fibrosis, or cardiac fibrosis in a cell, tissue or subject in need thereof, comprising administering a therapeutically effective amount of a compound of Formula (1), (2), (3), (4), (5), (5 A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B), or a pharmaceutically acceptable salt thereof to said subject.
Pulmonary Fibrosis
[0098] LPA is an important mediator of fibroblast recruitment in pulmonary fibrosis. LPA and LPA1 play key pathogenic roles in pulmonary fibrosis. Fibroblast chemo-attractant activity plays an important role in the lungs in patients with pulmonary fibrosis. Profibrotic effects of LPA1 -receptor stimulation is explained by LPA1 -receptor-mediated vascular leakage and increased fibroblast recruitment, both profibrotic events. The LPA-LPA1 pathway has a role in mediating fibroblast migration and vascular leakage in IPF. The end result is the aberrant healing process that characterizes this fibrotic condition.
[0099] The LPA1 receptor is the LPA receptor most highly expressed on fibroblasts obtained from patients with IPF. Furthermore, bronchoalveolar lavage (BAL) obtained from IPF patients induced chemotaxis of human fetal lung fibroblasts that was blocked by the dual LPA1- LP A3 receptor antagonist Ki 16425. In an experimental bleomycin- induced lung injury mouse model, it was shown that LPA levels were high in BAL samples compared with unexposed controls. LPA1 knockout mice are protected from fibrosis after bleomycin challenge with reduced fibroblast accumulation and vascular leakage. In human subjects with IPF, high LPA levels were observed in bronchoalveolar lavage samples compared with healthy controls. Increased fibroblast chemotactic activity in these samples was inhibited by the Ki 16425, indicating that fibroblast migration is mediated by the LPA- LPA receptor(s) pathway (Tager et al. Nature Medicine, vol. 14, no. 1, 45-54, 2008). A recent study also showed that a LPA1 receptor antagonist inhibits lung fibrosis in a mouse bleomycin model (Swaney et al., Brit. J. Pharmacol, vol. 160, 1699-1713 (2010)). The LPA-LPA1 pathway also plays an important role in fibroblast recruitment and vascular leakage in pulmonary fibrosis.
Renal Fibrosis
[0100] LPA and LPA1 are involved in the etiology of kidney fibrosis. LPA has effects on both proliferation and contraction of glomerular mesangial cells and thus has been implicated in proliferative glomerulonephritis (Inoue et al., Clinical Science 96, 431- 436, 1999).
[0101] In an animal model of renal fibrosis, unilateral ureteral obstruction (UUO), it was found that renal LPA receptors are expressed under basal conditions with an expression order of LPA2>LPA3=LPA1 >> LPA4. This model mimics in an accelerated manner the development of renal fibrosis including renal inflammation, fibroblast activation and accumulation of extracellular matrix in the tubulointerstitium. UUO significantly induced LPAl-receptor
expression. This was paralleled by renal LPA production (3.3 fold increase) in conditioned media from kidney explants. Contra-lateral kidneys exhibited no significant changes in LPA release and LPA-receptors expression. This shows that a prerequisite for an action of LPA in fibrosis is met: production of a ligand (LPA) and induction of one of its receptors (the LPA1 receptor) (J.P. Pradere et al., Biochimica et Biophysica Acta, Molecular and Cell Biology of Lipids (2008), 1781(9), 582-587). In mice invalidated for the LPA1 receptor (LPA1 (T), the development of renal fibrosis was significantly attenuated. UUO mice treated with the LPA receptor antagonist Kil6425 closely resembled the LPA1 (7 ) mice.
[0102] LPA can participate in intraperitonial accumulation of monocyte/macrophages and that LPA can induce expression of the profibrotic cytokine CTGF in primary cultures of human fibroblasts (Koh et al., J. Clin. Invest. 102 (1998) 716-727). LPA treatment of a mouse epithelial renal cell line, MCT, induced a rapid increase in the expression of the profibrotic cytokine CTGF. CTGF plays a crucial role in UUO- induced tubulointerstitial fibrosis (TIF), and is involved in the profibrotic activity of TGF beta. This induction was almost completely suppressed by co-treatment with the LPA-receptor antagonist Kil6425. In one aspect, the profibrotic activity of LPA in kidney results from a direct action of LPA on kidney cells involving induction of CTGF.
Hepatic fibrosis
[0103] LPA is implicated in liver disease and fibrosis. Plasma LPA levels and serum autotoxin (enzyme responsible for LPA production) are elevated in hepatitis patients and animal models of liver injury in correlation with increased fibrosis. LPA also regulates liver cell function. LPA1 and LPA2 receptors are expressed by mouse hepatic stellate cells and LPA stimulates migration of hepatic myofibroblasts.
Ocular Fibrosis
[0104] LPA is in involved in wound healing in the eye. LPA1 and LPA3 receptors are detectable in the normal rabbit corneal epithelial cells, keratocytes and endothelial cells and LPA1 and LPA3 expression are increased in corneal epithelial cells following injury. LPA and its homologues are present in the aqueous humor and the lacrimal gland fluid of the rabbit eye and these levels are increased in a rabbit corneal injury model. LPA induces actin stress fiber formation in rabbit corneal endothelial and epithelial cells and promotes contraction corneal fibroblasts. LPA also stimulates proliferation of human retinal pigmented epithelial cells
Cardiac fibrosis
[0105] LPA is implicated in myocardial infarction and cardiac fibrosis. Serum LPA levels are increased in patients following myocardial infarction (MI) and LPA stimulates proliferation and collagen production (fibrosis) by rat cardiac fibroblasts. Both LPAland LPA3 receptors are highly expressed in human heart tissue.
Administration and Pharmaceutical Compositions
[0106] In another aspect, the present invention provides a pharmaceutical composition comprising a compound of the present invention or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier. The pharmaceutical composition can be formulated for particular routes of administration such as oral administration, parenteral administration, and rectal administration, etc. In addition, the pharmaceutical compositions of the present invention can be made up in a solid form (including without limitation capsules, tablets, pills, granules, powders or suppositories), or in a liquid form (including without limitation solutions, suspensions or emulsions). The pharmaceutical compositions can be subjected to conventional pharmaceutical operations such as sterilization and/or can contain conventional inert diluents, lubricating agents, or buffering agents, as well as adjuvants, such as preservatives, stabilizers, wetting agents, emulsifers and buffers, etc.
[0107] Typically, the pharmaceutical compositions are tablets or gelatin capsules comprising the active ingredient together with
a) diluents, e.g., lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycine; b) lubricants, e.g., silica, talcum, stearic acid, its magnesium or calcium salt and/or poly ethylenegly col; for tablets also
c) binders, e.g. , magnesium aluminum silicate, starch paste, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone; if desired
d) disintegrants, e.g. , starches, agar, alginic acid or its sodium salt, or effervescent mixtures; and/or
e) absorbents, colorants, flavors and sweeteners.
Tablets may be either film coated or enteric coated according to methods known in the art.
[0108] Suitable compositions for oral administration include an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof, in the form of tablets, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft
capsules, or syrups or elixirs. Compositions intended for oral use are prepared according to any method known in the art for the manufacture of pharmaceutical compositions and such compositions can contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide
pharmaceutically elegant and palatable preparations. Tablets may contain the active ingredient in admixture with nontoxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets. These excipients are, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example, starch, gelatin or acacia; and lubricating agents, for example magnesium stearate, stearic acid or talc. The tablets are uncoated or coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate can be employed. Formulations for oral use can be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example, peanut oil, liquid paraffin or olive oil.
[0109] Certain injectable compositions are aqueous isotonic solutions or suspensions, and suppositories are advantageously prepared from fatty emulsions or suspensions. Said compositions may be sterilized and/or contain adjuvants, such as preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers. In addition, they may also contain other therapeutically valuable substances. Said compositions are prepared according to conventional mixing, granulating or coating methods, respectively, and contain about 0.1-75%, or contain about 1-50%, of the active ingredient.
[0110] Suitable compositions for transdermal application include an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof, with a suitable carrier. Carriers suitable for transdermal delivery include absorbable pharmacologically acceptable solvents to assist passage through the skin of the host. For example, transdermal devices are in the form of a bandage comprising a backing member, a reservoir containing the compound optionally with carriers, optionally a rate controlling barrier to deliver the compound of the skin of the host at a controlled and predetermined rate over a prolonged period of time, and means to secure the device to the skin.
[0111] Suitable compositions for topical application, e.g. , to the skin and eyes, include aqueous solutions, suspensions, ointments, creams, gels or sprayable formulations, e.g. , for delivery by aerosol or the like. Such topical delivery systems will in particular be appropriate for dermal application, e.g. , for the treatment of skin cancer, e.g. , for prophylactic use in sun creams, lotions, sprays and the like. They are thus particularly suited for use in topical, including cosmetic, formulations well-known in the art. Such may contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
[0112] As used herein, a topical application may also pertain to an inhalation or to an intranasal application. They may be conveniently delivered in the form of a dry powder (either alone, as a mixture, for example a dry blend with lactose, or a mixed component particle, for example with phospholipids) from a dry powder inhaler or an aerosol spray presentation from a pressurised container, pump, spray, atomizer or nebuliser, with or without the use of a suitable propellant.
[0113] Dosage forms for the topical or transdermal administration of a compound of this invention include powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches and inhalants. The active compound may be mixed under sterile conditions with a
pharmaceutically acceptable carrier, and with any preservatives, buffers, or propellants that may be desirable.
[0114] The ointments, pastes, creams and gels may contain, in addition to an active compound of this invention, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
[0115] Powders and sprays can contain, in addition to a compound of this invention, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances. Sprays can additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
[0116] Transdermal patches have the added advantage of providing controlled delivery of a compound of the present invention to the body. Such dosage forms can be made by dissolving or dispersing the compound in the proper medium. Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate of such flux can be controlled by either providing a rate controlling membrane or dispersing the active compound in a polymer
matrix or gel.
[0117] Ophthalmic formulations, eye ointments, powders, solutions and the like, are also contemplated as being within the scope of this invention.
[0118] The present invention further provides anhydrous pharmaceutical compositions and dosage forms comprising the compounds of the present invention or a pharmaceutically acceptable salt thereof as active ingredients, since water may facilitate the degradation of certain compounds. Anhydrous pharmaceutical compositions and dosage forms of the invention can be prepared using anhydrous or low moisture containing ingredients and low moisture or low humidity conditions. An anhydrous pharmaceutical composition may be prepared and stored such that its anhydrous nature is maintained. Accordingly, anhydrous compositions are packaged using materials known to prevent exposure to water such that they can be included in suitable formulary kits. Examples of suitable packaging include, but are not limited to, hermetically sealed foils, plastics, unit dose containers (e. g. , vials), blister packs, and strip packs.
[0119] The invention further provides pharmaceutical compositions and dosage forms that comprise one or more agents that reduce the rate by which the compound of the present invention as an active ingredient will decompose. Such agents, which are referred to herein as "stabilizers," include, but are not limited to, antioxidants such as ascorbic acid, pH buffers, or salt buffers, etc.
[0120] The pharmaceutical composition or combination of the present invention can be in unit dosage of about 1-1000 mg of active ingredient(s) for a subject of about 50-70 kg, or about 1-500 mg or about 1-250 mg or about 1-150 mg or about 0.5-100 mg, or about 1-50 mg of active ingredients. The therapeutically effective dosage of a compound, the pharmaceutical composition, or the combinations thereof, is dependent on the species of the subject, the body weight, age and individual condition, the disorder or disease or the severity thereof being treated. A physician, clinician or veterinarian of ordinary skill can readily determine the effective amount of each of the active ingredients necessary to prevent, treat or inhibit the progress of the disorder or disease.
[0121] The above-cited dosage properties are demonstrable in vitro and in vivo tests using advantageously mammals, e.g. , mice, rats, dogs, monkeys or isolated organs, tissues and preparations thereof. The compounds of the present invention or a pharmaceutically acceptable salt thereof can be applied in vitro in the form of solutions, e.g. , aqueous solutions, and in vivo
either enterally, parenterally, advantageously intravenously, e.g. , as a suspension or in aqueous solution. The dosage in vitro may range between about 10~3 molar and 10~9 molar
concentrations. A therapeutically effective amount in vivo may range depending on the route of administration, between about 0.1-500 mg/kg, or between about 1-100 mg/kg.
[0122] The compound of the present invention or a pharmaceutically acceptable salt thereof may be administered either simultaneously with, or before or after, one or more other therapeutic agent. The compound of the present invention may be administered separately, by the same or different route of administration, or together in the same pharmaceutical composition as the other agents.
[0123] In one embodiment, the invention provides a product comprising a compound of Formula (1), (2), (3), (4), (5), (5A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B) or a pharmaceutically acceptable salt thereof, and at least one other therapeutic agent as a combined preparation for simultaneous, separate or sequential use in the treatment of a disease or condition mediated by LPA. Products provided as a combined preparation include: a composition comprising a compound of the invention or a pharmaceutically acceptable salt thereof, and the other therapeutic agent(s) together in the same pharmaceutical composition; or a compound of the invention or a pharmaceutically acceptable salt thereof, and the other therapeutic agent(s) in separate form, e.g. in the form of a kit. Optionally, the pharmaceutical composition may comprise a pharmaceutically acceptable excipient, as described above.
[0124] In another embodiment, the invention provides a combination of a compound of the invention or a pharmaceutically acceptable salt thereof, and one or more additional
therapeutically active agents selected from: corticosteroids, immunosuppresants, analgesics, anti-cancer agent, anti-inflammatories, chemokine receptor antagonists, bronchodilators, leukotriene receptor antagonists, leukotriene formation inhibitors, monoacylglycerol kinase inhibitors, phospholipase Al inhibitors, phospholipase A2 inhibitors, and lysophospholipase D (lysoPLD) inhibitors, autotaxin inhibitors, decongestants, antihistamines, mucolytics, anticholinergics, antitussives, expectorants, and β-2 agonists.
[0125] In yet another embodiment, the invention provides a kit comprising two or more separate pharmaceutical compositions, at least one of which contains a compound of the invention or a pharmaceutically acceptable salt thereof. In one embodiment, the kit comprises means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet. An example of such a kit is a blister pack, as typically used for the packaging of
tablets, capsules and the like. The kit of the invention may be used for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another. To assist compliance, the kit of the invention typically comprises directions for administration.
[0126] In the combination therapies of the invention, the compound of the invention or a pharmaceutically acceptable salt thereof and the other therapeutic agent may be manufactured and/or formulated by the same or different manufacturers. Moreover, the compound of the invention and the other therapeutic may be brought together into a combination therapy: (i) prior to release of the combination product to physicians (e.g. in the case of a kit comprising the compound of the invention and the other therapeutic agent); (ii) by the physician themselves (or under the guidance of the physician) shortly before administration; (iii) in the patient themselves, e.g. during sequential administration of the compound of the invention and the other therapeutic agent.
[0127] Accordingly, the invention provides the use of a compound of the invention (e.g., Formula (1), (2), (3), (4), (5), (5A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B)) or a pharmaceutically acceptable salt thereof, for treating a disease or condition mediated by LPA, wherein the medicament is prepared for administration with another therapeutic agent. The invention also provides the use of another therapeutic agent for treating a disease or condition mediated by LPA, wherein the medicament is administered with a compound of the invention or a pharmaceutically acceptable salt thereof.
[0128] The invention also provides a compound of the invention for use in a method of treating a disease or condition mediated by LPA, wherein a compound of the invention (e.g., Formula (1), (2), (3), (4), (5), (5A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B)) or a pharmaceutically acceptable salt thereof, is prepared for administration with another therapeutic agent. The invention also provides another therapeutic agent for use in a method of treating a disease or condition mediated by LPA, wherein the other therapeutic agent is prepared for administration with a compound of the invention. The invention also provides a compound of the invention, for use in a method of treating a disease or condition mediated by LPA, wherein a compound of the invention or a pharmaceutically acceptable salt thereof is administered with another therapeutic agent. The invention also provides another therapeutic agent for use in a method of treating a disease or condition mediated by LPA, wherein the other therapeutic agent is administered with a compound of the invention.
[0129] The invention also provides the use of a compound of the invention (e.g., Formula (1), (2), (3), (4), (5), (5A), (5'), (5B), (5C), (6), (7), (8), (9A), (9B), (9C), (9D), (10A) or (10B)) or a pharmaceutically acceptable salt thereof, for treating a disease or condition mediated by LPA, wherein the patient has previously (e.g. within 24 hours) been treated with another therapeutic agent. The invention also provides the use of another therapeutic agent for treating a disease or condition mediated by LPA, wherein the patient has previously (e.g. within 24 hours) been treated with a compound of the invention.
Processes for Making Compounds of the Invention
[0130] Typically, the compounds of the invention can be prepared according to any one of Schemes 1, 2, 3 or 4, provided infra. Each reaction step can be carried out in a manner known to those skilled in the art. For example, a reaction can be carried out in the presence of a suitable solvent or diluent or of mixture thereof. A reaction can also be carried out, if needed, in the presence of an acid or a base, with cooling or heating, for example in a temperature range from approximately -30 °C to approximately 150 °C. In particular examples, a reaction is carried out in a temperature range from approximately 0 °C to 100 °C, and more particularly, in a temperature range from room temperature to approximately 80 °C, in an open or closed reaction vessel and/or in the atmosphere of an inert gas, for example nitrogen.
[0131] Typically, compounds of Formula (1) can be prepared following the procedures in Scheme 1:

Scheme 1
wherein Z1, Z2, R1, R2, R3, R4a, R4b, R5, R6, R6a ,R6b, B, m, n and p are as previously defined
1 2 20 20 in Formula (1); X, X , and X are leaving groups; and R is hydrogen or Ci^alkyl; or two R groups together with boron can form a cyclic boronate ester.
[0132] A compound of Formula (1) can be prepared by Suzuki reaction of a compound of Formula 1-4 with a compound of Formula 1-5, as described in detail below in Scheme 2a. A compound of Formula 1-4 can, in turn, be prepared by reaction of a compound of Formula 1-1 with an alkoxycarbonylation reagent of Formula 1-3. Suitable alkoxycarbonylation reagents include chloroformates and dialkyl dicarbonates (for example, di-tert-butyl dicarbonate). The reaction generally takes place in the presence of a base such as triethylamine or pyridine, in an inert solvent such as dichloromethane or dioxane, at a temperature of about -78 °C to about rt.
Alternatively, the reaction can take place in a biphasic system consisting of an inert solvent such as dioxane together with an aqueous basic solution such as aqueous sodium carbonate or sodium bicarbonate.
[0133] A compound of Formula 1-4 can also be prepared by reaction of a compound of Formula 1-1 with phosgene, followed by treatment with an alcohol of Formula 1-2. A compound of Formula (1) can also be prepared by reversal of the order of the above reaction steps; that is, Suzuki reaction of a compound of Formula 1-1 with a compound of Formula 1-5, followed by reaction of the resulting compound of Formula 1-6 with phosgene and an alcohol of Formula 1-2. An alternative preparation of an intermediate compound of Formula 1-6 can be performed by Suzuki reaction of a compound of Formula 1-8 with a boronic acid derivative of Formula 1-9. A compound of Formula 1-8 can in turn be prepared by selective Suzuki reaction of a compound of Formula 1-1 with a halophenylboronic acid derivative (preferably a chlorophenylboronic acid derivative in which X1 is CI) of Formula 1-7.
[0134] A compound of Formula (1) can also be prepared by Suzuki reaction of a compound of Formulal-9 with a compound of Formula 1-11. A compound of Formula 1-11 can in turn be prepared by selective Suzuki reaction of a compound of Formula 1-4 with a halophenylboronic acid derivative (preferably a chlorophenylboronic acid derivative in which X1 is CI) of Formula 1-7. Alternatively, a compound of Formula 1 can be prepared by a Suzuki reaction in which the roles of the coupling partners are reversed; that is, by reaction of a boronic acid derivative of Formula 1-12 with a compound of Formula 1-13. A compound of Formula 1-12 can be prepared by treatment of a compound of Formula 1-11 with a diboron compound (for example, bis- pinacolatodiboron) under palladium catalysis.
[0135] A compound of Formula (1) can also be prepared by reaction of a compound of Formula 1-16 with DPP A, in the presence of a compound of Formula 1-2 and a base. A compound of Formula 1-16 can in turn be prepared by Suzuki coupling of a compound of Formula 1-15 with a compound of Formula 1-9, followed by ester hydrolysis. A compound of Formula 1-15 can be prepared by cycloaddition reaction of a compound of Formula 1-14 with a diazoacetate ester, followed by alkylation with an alkylating agent R5X, in the presence of a base such as sodium hydride.
[0136] A compound of Formula I- 11 can also be prepared by ester hydrolysis of a compound of Formula 1-15, followed by reaction with DPPA, in the presence of a compound of Formula 1-2 and a base.
[0137] Compounds of Formula 1-1, 1-2, 1-3, 1-5, 1-7, 1-9, 1-13, and 1-14 are commercially available, or can be prepared by literature methods or by methods available to those skilled in the art.
[0138] In one embodiment, compounds of Formula (2) can be prepared following the

base wherein Z2, R1, R2, R3, R4a, R4b, R5, R6, R7, R8, B, m, n and p are as previously defined in
1 20
Formula (1); X and X are leaving groups; and R is as described in Scheme 1.
[0139] In Scheme 2a, compounds of Formula (2) can be prepared by coupling a compound of Formula 2a-2, in which X is a leaving group (e.g., iodo, bromo, chloro, trifluoromethane- sulfonyloxy, and the like), with a compound of Formula 1-5. The reaction takes place in the presence of a suitable transition metal catalyst (e.g., tetrakis(triphenylphosphinepalladium)(0), PdCl2(dppf)), or dichloro [Ι,Γ bis(di-tert- butylphosphino)]ferrocene palladium (II)), a suitable solvent (e.g., DME, dioxane, toluene, ethanol, and the like) and a suitable base (e.g., anhydrous potassium carbonate or aqueous sodium carbonate solution, and the like). The reaction proceeds
in a temperature range of about 20°C to about 120°C and can take up to about 24 hours to complete. The reaction mixture is optionally further reacted to remove any protecting groups. It will be appreciated by one skilled in the art that other organometallic coupling reactions, for example using tin reagents (Stille coupling) or zinc reagents (Negishi coupling), may also be employed in place of the Suzuki coupling reaction using boron reagents described in Scheme 2a.
[0140] Intermediate compounds of Formula 2a-2 can be prepared by a reaction of a compound of Formula 2-1 with phosgene, followed by reaction with an alcohol of Formula 2a- 1. The reaction takes place in the presence of a suitable base such as pyridine, optionally in the presence of a drying agent such as molecular sieves, in an inert solvent such as dichloromethane, THF, and the like, at a temperature from about 20 °C to about 65 °C.
[0141] Alternatively, compounds of Formula (2) can be prepared by an alternate sequence in which the order of reaction steps is reversed. Thus, Suzuki reaction of a compound of Formula 2-1 with a compound of Formula 1-5 provides an intermediate compound of Formula 1-6, which can then be reacted with DPPA and a compound of Formula 2a- 1 as described above to furnish a compound of Formula (2). Intermediate compounds of Formula 1-6 can also be prepared by Suzuki reaction of a compound of Formula 1-9 with a compound of Formula 2-8.
[0142] In another embodiment, compounds of Formula (2) can be prepared following the procedures in Scheme 3:

1 2 20
Formula (1); X and X are leaving groups; and R is as described in Scheme 1.
[0143] Compounds of Formula (2) can be prepared by Suzuki coupling of a compound of Formula 2b- 1 with a compound of Formula 1-9, using similar methods to those described in Scheme 2a. Compounds of Formula (2) can also be prepared by Suzuki reaction in which the roles of the coupling partners are reversed; that is, by reaction of a boronic acid derivative of Formula 2b-2 with a compound of Formula 1-13. A compound of Formula 2b-2 can be prepared by treatment of a compound of Formula 2b- 1 with a diboron compound (for example, bis(pinacolato)diboron and the like) in the presence of a suitable transition metal catalyst (for example PdCl2(dppf)),) and a suitable base (for example, potassium acetate and the like) in a suitable solvent (for example, toluene, dioxane and the like). The reaction proceeds in a temperature range of about 20 °C to about 120 °C and can take up to about 24 hours to complete.
[0144] In another embodiment, compounds of Formula (2) or Formula 2b-2 can be prepared as describe

Scheme 4
 for compounds of Formula (2); or E is OH, OP, Br, CI, or I, wherein P is a protecting group, for compounds of Formula 2b- 1 ; and
for compounds of Formula (2); or E is OH, OP, Br, CI, or I, wherein P is a protecting group, for compounds of Formula 2b- 1 ; and
Z2, R1, R2, R3, R4a, R4b, R5, R6, R7, R8, B, m, n and p are as previously defined in Formula (1).
[0145] In Scheme 4, compounds of Formula (2) or 2b- 1 can be prepared by reaction of a carboxylic acid compound of Formula 4- 1 with diphenylphosphoryl azide (DPP A) and an alcohol of Formula 2a- 1, in an inert solvent such as dichloromethane, in the presence of a base
such as triethylamine or DIPEA. If desired, a compound of Formula (2) can be converted to another compound of Formula (2), or a compound of Formula 2b- 1 can be converted to another compound of Formula 2b- 1.
Intermediate Compounds
[0146] Intermediate compounds of Formula 2- 1 in Scheme 2a can be prepared following literature methods or by following the procedures in Scheme 5:

2_i Scheme 5 wherein R5 is as previously defined in Formula (2); and X and X' are leaving groups.
[0147] In Scheme 5, compounds of Formula 2-1 can be prepared by halogenation of a pyrazole compound of Formula 2- lb. When X is bromine, appropriate halogenating agents include N-bromosuccinimide and Br2; when X is iodine, appropriate halogenating agents include N-iodosuccinimide; and when X is chlorine, appropriate reagents include N-chlorosuccinimide and Cl2. The reaction takes place in a suitable solvent such as dichloromethane, at a temperature from about -78 °C to about 40 °C. A compound of Formula 2- lb can be prepared by reaction of a compound of Formula 2- la with an alkylating agent R5X' , in which X' is a halogen such as chloro, bromo, or iodo, or an alkyl- or aryl-sulfonyloxy group. The alkylation takes place in the presence of a suitable base such as sodium hydride, potassium carbonate, or the like, in a suitable solvent such as DMF or DMSO, at a temperature of about 0 °C to about 120 °C. During this reaction the isomeric products of Formulas 2-lc and 2-ld may be formed. Compounds of Formulas 2- lb can be separated from isomers 2-lc and 2-ld by those skilled in the art using techniques such as silica gel chromatography, crystallization, or reverse-phase preparative HPLC.
[0148] An intermediate compound of Formula 6-3 is a particular embodiment of a compound of Formula 2-8 in which X1 is CI. Such compounds can be prepared as shown in
Scheme 6:

Scheme 6
[0149] In Scheme 6, a compound Formula 6-3, can be prepared by coupling a compound of Formula 6-1 (i.e., a compound of Formula 1-1 in which X is Br and Z1 is CH), with a compound of Formula 6-2. The reaction takes place in the presence of a suitable transition metal catalyst (e.g., tetrakis(triphenylphosphinepalladium)(0), PdCi2(dppf)), or dichloro [Ι, bis(di-tert- butylphosphino)]ferrocene palladium (II)), a suitable solvent (e.g., DME, dioxane, toluene, ethanol, and the like) and a suitable base (e.g., anhydrous potassium carbonate or aqueous sodium carbonate solution, and the like). The reaction proceeds in a temperature range of about 20°C to about 120°C and can take up to about 24 hours to complete. The reaction mixture is optionally further reacted to remove any protecting groups. It will be appreciated by one skilled in the art that other organometallic coupling reactions, for example using tin reagents (Stille coupling) or zinc reagents (Negishi coupling), may also be employed in place of the boron reagents used in the Suzuki coupling reaction described in Scheme 6.
[0150] Intermediate compounds of Formula 2b- 1 in Scheme 3 can be prepared as described in Scheme 7:

X1 = CI Scheme 7 wherein Z2, R1, R2, R3, R4a, R4b, R5, R6, R7, R8, B, m, n and p are as previously defined in Formula (2); X and X1 are leaving groups; and R20 is as defined in Scheme 1.
[0151] In Scheme 7, compounds of Formula 2b- 1 in which X1 is CI, Br, or I can be prepared by Sandmeyer reaction of an aniline compound of Formula 7-3. The reaction is typically performed using sodium nitrite in acidic aqueous medium, or an alkyl nitrite in aqueous or nonaqueous solution, in the presence of a copper(I) or copper(II) halide salt. For example, the reaction can be performed using copper(II) bromide and an alkyl nitrite such as tert-butyl nitrite, in a suitable solvent such as anhydrous acetonitrile, at a temperature of about rt, for a time of about 1-4 h. A compound of Formula 2b- 1 in which X1 is triflate can be prepared by reaction of a compound of Formula 7-4 with trifluoromethanesulfonic anhydride, in an inert solvent such as dichloromethane, in the presence of a base such as N,N-dimethylaminopyridine or pyridine, at a temperature of about -78 °C to about rt. Compounds of Formula 7-3 and 7-4 can in turn be prepared by Suzuki coupling of a compound of Formula 7-1 or 7-2, respectively, with a compound of Formula 2a-2 as described earlier in Scheme 2a. For these reactions, the amino and hydroxyl groups may optionally be protected with a protecting group P. A compound of
Formula 2b- 1 in which X1 is CI can be prepared directly via Suzuki coupling of a compound of Formula 2a-2 with a chlorophenylboronate compound of Formula 6-2.
[0152] Intermediate reagents in Scheme 4 can be prepared following the procedures in Sc

wherein E is (R )n ; or E is OH, OP, Br, CI, or I, wherein P is a protecting group; and Z2, R1, R2, R3, R4a, R4b, R5, R6, R7, R8, B, m, n and p are as previously defined in Formula (2).
[0153] In Scheme 8, a carboxylic acid of Formula 4-1 can be prepared by hydrolysis of an ester compound of Formula 8-3a using acidic or basic methods known to those skilled in the art; for example, by treatment with lithium hydroxide in a mixture of THF and water, at about 0 °C to about 65 °C. An ester compound of Formula 8-3a (or an analogous lower alkyl ester) can be prepared via alkylation of a pyrazole compound of Formula 8-2 with an alkylating agent R5X', in which X' is a halogen such as chloro, bromo, or iodo, or an alkyl- or aryl-sulfonyloxy group. The alkylation takes place in the presence of a suitable base such as sodium hydride, potassium carbonate, or the like, in a suitable solvent such as DMF or DMSO, at a temperature of about 0 °C to about 120 °C. During this reaction the isomeric product of Formula 8-3b may be formed. Compounds of Formulas 8-3a and 8-3b can be separated by those skilled in the art using
techniques such as silica gel chromatography, crystallization, or reverse-phase preparative HPLC. A compound of Formula 8-2 can be prepared by reaction of a nitroalkene compound of Formula 8-1 with a diazoacetate ester such as ethyl diazoacetate. The reaction takes place in an inert solvent such as THF or toluene, at a temperature of about 20 °C to about 150 °C.
[0154] Alternatively, intermediate compounds of Formula 8-2 can be prepared by reaction of an alkyne compound of Formula 8-4 with a diazoacetate ester such as ethyl diazoacetate. The reaction takes place in an inert solvent such as THF or toluene, at a temperature of about 20 °C to about 150 °C. During this reaction the isomeric product of Formula 8-5 may be formed. Compounds of Formulas 8-2 and 8-5 can be separated by those skilled in the art using techniques such as silica gel chromatography, crystallization, or reverse-phase preparative HPLC.
[0155] Intermediate compounds of Formula 9-3 can be prepared following the procedures in Scheme

Scheme 9
3 4b 20 2 wherein R , R , R , and m are as previously defined in Formula (1); and X is CI, Br, I.
[0156] In Scheme 9, a compound of Formula 9-3 can be prepared by reaction of a compound of Formula 9-2 with a diboron compound (for example, bis(pinacolato)diboron and the like) in the presence of a suitable transition metal catalyst (for example PdCl2(dppf)),) and a suitable base (for example, potassium acetate and the like) in a suitable solvent (for example, toluene, dioxane and the like). The reaction proceeds in a temperature range of about 20 °C to about 120 °C and can take up to about 24 hours to complete. A compound of Formula 9-2 can in turn be prepared by alkylation of a compound of Formula 9-1 with a bis-electrophile (for example, 1,2- dibromoe thane). The reaction takes place in the presence of a suitable base (for example, sodium hydride) in a solvent such as DMF, at a temperature of about 0 °C to about 100 °C. Alternatively, the reaction can take place under phase transfer conditions using an organic solvent such as toluene or dichloromethane, an aqueous base such as sodium or potassium hydroxide solution, and a phase transfer agent such as tetrabutylammonium bromide, at a temperature of about 0 °C to about 50 °C.
[0157] Typically, compounds of Formula 10-10 can be prepared following the procedures in Scheme 10:
X X Hal
Halogenation 1 Methylation X1 Hal 1. BuLi, THF
HN, 7
N=N HN> 7 2. DMF
e.g. Br2 N=N e.g. Me-I, base N-N N-N
10-1 10-2 10-3 /
10-4

Scheme 10
wherein R1, R2, R3, R4a, R4b, R5, R6, R6a, R6b, B, m, n and p are as previously defined in
1 20
Formula (1); each X is a leaving group; and R is as defined in Scheme 1.
[0158] In scheme 10, a compound of Formula 10-10 can be prepared by a metal-catalyzed cross-coupling reaction of a compound of Formula 10-9 with a boronic acid such as a substituted phenyl boronic acid of Formula 1-9, in the presence of a palladium catalyst such as dichloro [Ι,Γ bis(di-tert-butylphosphino)]ferrocene palladium (II), a base such as potassium phosphate tribasic in an inert solvent such as 1 ,4-dioxane with heating at elevated temperatures either thermally or with microwave irradiation. A person skilled in the art would understand that other suitable palladium catalysts, bases and solvents may be used.
[0159] Intermediate compounds of Formula 10-9 can be prepared by Curtis rearrangement of a carboxylic acid of Formula 10-7 in the presence of an alcohol of Formula 10-8, following analogous procedures as described in Scheme 4. Intermediate compounds of Formula 10-7 can be prepared by oxidation of compounds of Formula 10-6 for example using Oxone in DMF. A person skilled in the art would understand that other suitable oxidizing agents and solvents may be used.
[0160] Intermediate compounds of Formula 10-6 can be prepared by a metal-catalyzed cross-coupling reaction of a compound of Formula 10-5 with a boronic acid such as a substituted phenyl boronic acid of Formula 1-7, in which X1 is chloro, in the presence of a palladium catalyst such as dichloro [Ι,Γ bis(di-tert-butylphosphino)]ferrocene palladium (II), a base such as potassium phosphate tribasic in an inert solvent such as 1,4-dioxane with heating at elevated temperatures either thermally or with microwave irradiation. A person skilled in the art would understand that other suitable palladium catalysts, bases and solvents may be used.
[0161] Intermediate compounds of Formula 10-5 can be prepared by a lithium-halogen exchange of a compound of Formula 10-3 with an alkyllithium reagent (such as butyllithium) followed by reaction with a formylating reagent such as DMF, or N-formylmorpholine or ethyl formate in a suitable solvent such as THF, at a temperature from about -78 °C to 40 °C.
Compounds of Formula 10-3 can in turn be prepared by a methylation reaction of a compound of Formula 10-2 in the presence of a base such as potassium carbonate, suitable methylating agents include iodomethane, dimethylsulfate, methyl triflate, methyl mesylate in a suitable solvent such as THF, at a temperature from about -20 °C to 60 °C. In the course of this reaction regioisomer 10-4 may also be formed. A person skilled in the art would understand that other suitable methylating agents, bases and solvents may be used.
[0162] Intermediate compounds of Formula 10-2 can be prepared by halogenation of a triazole of Formula 10-1. When X is bromine, appropriate halogenating agents include N- bromosuccinimide and Br2; when X is iodine, appropriate halogenating agents include N- iodosuccinimide and iodine; and when X is chlorine, appropriate reagents include N- chlorosuccinimide and Cl2. The reaction takes place in a suitable solvent such as
dichloromethane, at a temperature from about -78 °C to about 40 °C. A person skilled in the art would understand that other suitable halogenating agents and solvents may be used. It will be appreciated by one skilled in the art that other organometallic coupling reactions, for example using tin reagents (Stille coupling) or zinc reagents (Negishi coupling), may also be employed in place of the boron reagents used in the Suzuki coupling reaction described in Scheme 10.
[0163] The invention also relates to those forms of the process in which a compound obtainable as an intermediate at any stage of the process is used as starting material and the remaining process steps are carried out, or in which a starting material is formed under the reaction conditions or is used in the form of a derivative, for example in a protected form or in the form of a salt, or a compound obtainable by the process according to the invention is
produced under the process conditions and processed further in situ. Compounds of the invention and intermediates can also be converted into each other according to methods generally known to those skilled in the art. Intermediates and final products can be worked up and/or purified according to standard methods, e.g. using chromatographic methods, distribution methods, (re-) crystallization, and the like.
[0164] Within the scope of this text, only a readily removable group that is not a constituent of the particular desired end product of the compounds of the present invention is designated a "protecting group", unless the context indicates otherwise. The protection of functional groups by such protecting groups, the protecting groups themselves, and their cleavage reactions are described for example in standard reference works, such as J. F. W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973; in T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999; in "The Peptides"; Volume 3 (editors: E. Gross and J. Meienhofer), Academic Press, London and New York 1981; in "Methoden der organischen Chemie" (Methods of Organic Chemistry), Houben Weyl, 4th edition, Volume 15/1, Georg Thieme Verlag, Stuttgart 1974; in H.-D. Jakubke and H. Jeschkeit, "Aminosauren, Peptide, Proteine" (Amino acids, Peptides, Proteins), Verlag Chemie, Weinheim, Deerfield Beach, and Basel 1982; and in Jochen
Lehmann, "Chemie der Kohlenhydrate: Monosaccharide und Derivate" (Chemistry of
Carbohydrates: Monosaccharides and Derivatives), Georg Thieme Verlag, Stuttgart 1974; and in subsequent versions thereof. A characteristic of protecting groups is that they can be removed readily (i.e. without the occurrence of undesired secondary reactions) for example by solvolysis, reduction, photolysis or alternatively under physiological conditions (e.g. by enzymatic cleavage).
[0165] All the above-mentioned process steps mentioned herein before and hereinafter can be carried out under reaction conditions that are known to those skilled in the art, including those mentioned specifically, in the absence or, customarily, in the presence of solvents or diluents, including, for example, solvents or diluents that are inert towards the reagents used and dissolve them, in the absence or presence of catalysts, condensation or neutralizing agents, for example ion exchangers, such as cation exchangers, e.g. in the H+ form, depending on the nature of the reaction and/or of the reactants at reduced, normal or elevated temperature, for example in a temperature range of from about -100 °C to about 190 °C, including, for example, from approximately -80 °C to approximately 150 °C, for example at from -80 to -60 °C, at room
temperature, at from -20 to 40 °C or at reflux temperature, under atmospheric pressure or in a closed vessel, where appropriate under pressure, and/or in an inert atmosphere, for example under an argon or nitrogen atmosphere.
[0166] At all stages of the reactions, mixtures of isomers that are formed can be separated into the individual isomers, for example diastereoisomers or enantiomers, or into any desired mixtures of isomers, for example racemates or mixtures of diastereoisomers. Mixtures of isomers obtainable according to the invention can be separated in a manner known to those skilled in the art into the individual isomers; diastereoisomers can be separated, for example, by partitioning between polyphasic solvent mixtures, recrystallisation and/or chromatographic separation, for example over silica gel or by e.g. medium pressure liquid chromatography over a reversed phase column, and racemates can be separated, for example, by the formation of salts with optically pure salt-forming reagents and separation of the mixture of diastereoisomers so obtainable, for example by means of fractional crystallisation, or by chromatography over optically active column materials.
[0167] The solvents from which those solvents that are suitable for any particular reaction may be selected include those mentioned specifically or, for example, water, esters, such as lower alkyl-lower alkanoates, for example ethyl acetate, ethers, such as aliphatic ethers, for example diethyl ether, or cyclic ethers, for example tetrahydrofuran or dioxane, liquid aromatic hydrocarbons, such as benzene or toluene, alcohols, such as methanol, ethanol or 1- or 2- propanol, nitriles, such as acetonitrile, halogenated hydrocarbons, such as methylene chloride or chloroform, acid amides, such as dimethylformamide or dimethyl acetamide, bases, such as heterocyclic nitrogen bases, for example pyridine or N-methylpyrrolidin-2-one, carboxylic acid anhydrides, such as lower alkanoic acid anhydrides, for example acetic anhydride, cyclic, linear or branched hydrocarbons, such as cyclohexane, hexane or isopentane, methycyclohexane, or mixtures of those solvents, for example aqueous solutions, unless otherwise indicated in the description of the processes. Such solvent mixtures may also be used in working up, for example by chromatography or partitioning.
[0168] The compounds of the present invention are either obtained in the free form, as a salt thereof, or as prodrug derivatives thereof. When both a basic group and an acid group are present in the same molecule, the compounds of the present invention may also form internal salts, e.g., zwitterionic molecules. In many cases, the compounds of the present invention are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl
groups or groups similar thereto. As used herein, the terms "salt" or "salts" refers to an acid addition or base addition salt of a compound of the invention. "Salts" include in particular "pharmaceutical acceptable salts". The term "pharmaceutically acceptable salts" refers to salts that retain the biological effectiveness and properties of the compounds of this invention and, which typically are not biologically or otherwise undesirable.
[0169] Salts of compounds of the present invention having at least one salt-forming group may be prepared in a manner known to those skilled in the art. For example, salts of compounds of the present invention having acid groups may be formed, for example, by treating the compounds with metal compounds, such as alkali metal salts of suitable organic carboxylic acids, e.g. the sodium salt of 2-ethylhexanoic acid, with organic alkali metal or alkaline earth metal compounds, such as the corresponding hydroxides, carbonates or hydrogen carbonates, such as sodium or potassium hydroxide, carbonate or hydrogen carbonate, with corresponding calcium compounds or with ammonia or a suitable organic amine, stoichiometric amounts or only a small excess of the salt-forming agent preferably being used. Acid addition salts of compounds of the present invention are obtained in customary manner, e.g. by treating the compounds with an acid or a suitable anion exchange reagent. Internal salts of compounds of the present invention containing acid and basic salt- forming groups, e.g. a free carboxy group and a free amino group, may be formed, e.g. by the neutralisation of salts, such as acid addition salts, to the isoelectric point, e.g. with weak bases, or by treatment with ion exchangers. Salts can be converted into the free compounds in accordance with methods known to those skilled in the art. Metal and ammonium salts can be converted, for example, by treatment with suitable acids, and acid addition salts, for example, by treatment with a suitable basic agent.
[0170] Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids, e.g., acetate, aspartate, benzoate, besylate, bromide/hydrobromide, bicarbonate/carbonate, bisulfate/sulfate, camphorsulfonate, chloride/hydrochloride,
chlorotheophyllinate, citrate, ethandisulfonate, fumarate, gluceptate, gluconate, glucuronate, hippurate, hydroiodide/iodide, isethionate, lactate, lactobionate, laurylsulfate, malate, maleate, malonate, mandelate, mesylate, methylsulphate, naphthoate, napsylate, nicotinate, nitrate, octadecanoate, oleate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, polygalacturonate, propionate, stearate, succinate, subsalicylate, tartrate, tosylate and trifluoroacetate salts.
[0171] Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
[0172] Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, sulfosalicylic acid, and the like. Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
[0173] Inorganic bases from which salts can be derived include, for example, ammonium salts and metals from columns I to XII of the periodic table. In certain embodiments, the salts are derived from sodium, potassium, ammonium, calcium, magnesium, iron, silver, zinc, and copper; particularly suitable salts include ammonium, potassium, sodium, calcium and magnesium salts.
[0174] Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like. Certain organic amines include isopropylamine, benzathine, cholinate, diethanolamine, diethylamine, lysine, meglumine, piperazine and tromethamine.
[0175] Other pharmaceutically acceptable salts can be derived from adipic acid (adipate), L- ascorbic acid (ascorbate), capric acid (caprate), sebacic acid (sebacate), l-hydroxy-2-naphthoic acid (xinafoate), L-glutamic acid (glutamate), glutaric acid (glutarate), triphenylacetic acid (trifenatate) and galactaric acid/mucic acid (mucate).
[0176] The pharmaceutically acceptable salts of the present invention can be synthesized from a parent compound, a basic or acidic moiety, by conventional chemical methods.
Generally, such salts can be prepared by reacting free acid forms of these compounds with a stoichiometric amount of the appropriate base (such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like), or by reacting free base forms of these compounds with a stoichiometric amount of the appropriate acid. Such reactions are typically carried out in water or in an organic solvent, or in a mixture of the two. Generally, use of non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile is desirable, where practicable. Lists of additional suitable salts can be found, e.g., in "Remington's Pharmaceutical Sciences", 20th ed., Mack Publishing Company, Easton, Pa., (1985); and in "Handbook of Pharmaceutical Salts:
Properties, Selection, and Use" by Stahl and Wermuth (Wiley- VCH, Weinheim, Germany,
2002), and in subsequent versions thereof.
[0177] The present invention also provides pro-drugs of the compounds of the present invention that converts in vivo to the compounds of the present invention. A pro-drug is an active or inactive compound that is modified chemically through in vivo physiological action, such as hydrolysis, metabolism and the like, into a compound of this invention following administration of the prodrug to a subject. The suitability and techniques involved in making and using pro-drugs are well known by those skilled in the art. Prodrugs can be conceptually divided into two non-exclusive categories, bioprecursor prodrugs and carrier prodrugs. See The Practice of Medicinal Chemistry, Ch. 31-32 (Ed. Wermuth, Academic Press, San Diego, Calif., 2001), and in subsequent versions thereof. Generally, bioprecursor prodrugs are compounds which are inactive or have low activity compared to the corresponding active drug compound; contain one or more protective groups, and are converted to an active form by metabolism or solvolysis. Both the active drug form and any released metabolic products should have acceptably low toxicity.
[0178] Carrier prodrugs are drug compounds that contain a transport moiety, e.g. , that improve uptake and/or localized delivery to a site(s) of action. Desirably for such a carrier prodrug, the linkage between the drug moiety and the transport moiety is a covalent bond, the prodrug is inactive or less active than the drug compound, and any released transport moiety is acceptably non-toxic. For prodrugs where the transport moiety is intended to enhance uptake, typically the release of the transport moiety should be rapid. In other cases, it is desirable to utilize a moiety that provides slow release, e.g. , certain polymers or other moieties, such as cyclodextrins. Carrier prodrugs can, for example, be used to improve one or more of the following properties: increased lipophilicity, increased duration of pharmacological effects, increased site-specificity, decreased toxicity and adverse reactions, and/or improvement in drug formulation (e.g. , stability, water solubility, suppression of an undesirable organoleptic or physiochemical property). For example, lipophilicity can be increased by esterification of (a) hydroxyl groups with lipophilic carboxylic acids (e.g., a carboxylic acid having at least one lipophilic moiety), or (b) carboxylic acid groups with lipophilic alcohols (e.g., an alcohol having at least one lipophilic moiety, for example aliphatic alcohols).
[0179] Exemplary prodrugs are, e.g. , esters of free carboxylic acids and 5-acyl derivatives of thiols and O-acyl derivatives of alcohols or phenols, wherein acyl has a meaning as defined herein. Suitable prodrugs are often pharmaceutically acceptable ester derivatives convertible by solvolysis under physiological conditions to the parent carboxylic acid, e.g. , lower alkyl esters, cycloalkyl esters, lower alkenyl esters, benzyl esters, mono- or di-substituted lower alkyl esters, such as the co-(amino, mono- or di-lower alkylamino, carboxy, lower alkoxycarbonyl)-lower alkyl esters, the oc-(lower alkanoyloxy, lower alkoxycarbonyl or di-lower alkylaminocarbonyl)- lower alkyl esters, such as the pivaloyloxymethyl ester and the like conventionally used in the art. In addition, amines have been masked as arylcarbonyloxymethyl substituted derivatives which are cleaved by esterases in vivo releasing the free drug and formaldehyde (Bundgaard, /. Med. Chem. 2503 (1989)). Moreover, drugs containing an acidic NH group, such as imidazole, imide, indole and the like, have been masked with N-acyloxymethyl groups (Bundgaard, Design of Prodrugs, Elsevier (1985)), and in subsequent versions thereof. Hydroxy groups have been masked as esters and ethers. EP 039,051 (Sloan and Little) discloses Mannich-base hydroxamic acid prodrugs, their preparation and use.
[0180] Furthermore, the compounds of the present invention, including their salts, may also be obtained in the form of hydrates, or their crystals may, for example, include the solvent used for crystallization. Different crystalline forms may be present. The compounds of the present invention may inherently or by design form solvates with pharmaceutically acceptable solvents (including water); therefore, it is intended that the invention embrace both solvated and unsolvated forms. The term "solvate" refers to a molecular complex of a compound of the present invention (including pharmaceutically acceptable salts thereof) with one or more solvent molecules. Such solvent molecules are those commonly used in the pharmaceutical art, which are known to be innocuous to the recipient, e.g., water, ethanol, and the like. The term "hydrate" refers to the complex where the solvent molecule is water. The compounds of the present invention, including salts, hydrates and solvates thereof, may inherently or by design form polymorphs.
[0181] Compounds of the invention in unoxidized form may be prepared from N-oxides of compounds of the invention by treating with a reducing agent (e.g., sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride, phosphorus trichloride, tribromide, or the like) in a suitable inert organic solvent (e.g. acetonitrile, ethanol, aqueous dioxane, or the like) at 0 to 80°C.
[0182] All starting materials, building blocks, reagents, acids, bases, dehydrating agents, solvents and catalysts utilized to synthesize the compounds of the present invention are either commercially available or can be produced by organic synthesis methods known to one of ordinary skill in the art (Houben-Weyl 4th Ed. 1952, Methods of Organic Synthesis, Thieme, Volume 21 , and subsequent versions thereof). All methods described herein can be performed in any suitable order unless otherwise indicated herein or otherwise clearly contradicted by context. The use of any and all examples, or exemplary language (e.g. "such as") provided herein is intended merely to better illuminate the invention and does not pose a limitation on the scope of the invention otherwise claimed.
Analytical Methods
[0183] The following HPLC and HPLC/MS methods were used in the preparation of Intermediates and Examples.
[0184] Method 1. Column: Waters Atlantis 2.1x30mm dC18
Mobile Phase: A) H20 + 0.5% TFA; B : ACN + 0.035% TFA
Pump method:

Detection: UV Diode Array at 214nm - 400nm
MS Scan: 160 - 800amu
ELSD: 50°C
[0185] Method 2. Column: Waters BEH C18 50x2.1 mm, 1.7 μιη; column temperature 50°C Mobile Phase: A) H20 + 0.1 % TFA; B: ACN + 0.1 % TFA
Pump method:
Flow
Time (min) A% B% (mL/min)
0-0.2 95 5 0.8 Isocratic
Linear
0.2 - 1.5 95^5 5^95 0.8 gradient
1.5-1.75 5 95 0.8 Isocratic
Detection: UV, MS
[0186] Method 3. Column: Waters BEH C18 50x2.1 mm, 1.7 μιη; column temperature 50°C Mobile Phase: A) H20 + 0.1% TFA; B: acetonitrile + 0.1% TFA
Pump method:

Detection: UV, MS
[0187] Method 4. Column: Waters CSH C18 100x2.1 mm, 1.7 μιη; column temperature°C
Mobile Phase: A) H20 + 0.1% formic; B: acetonitrile + 0.1% formic acid
Pump method:
Detection: UV, MS
Preparation of Intermediates
Intermediate 1
Ethyl 2-(4'-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)biphenyl-4-yl)acetate

KOAc, dioxane (INT-1 )
[0188] 2-(4'-Bromobiphenyl-4-yl)acetic acid. A mixture of l-(4'-bromobiphenyl-4- yl)ethanone (7.15 g, 0.026 mmol), sulfur (1.25 g, 0.039 mmol) and morpholine (20 ml) was heated at reflux for 4h. The mixture was concentrated and methanol (30 ml) was added. The mixture was sonicated and cooled, and the resulting solid (thioamide) was collected by filtration. To the solid was added a solution of NaOH (24 g) in water (120 ml) and ethanol (30 ml). The mixture was refluxed for 20h. The reaction was cooled, and the resulting precipitate was collected, washed with water and air-dried. The solid was then suspended in hot- water, and the mixture was acidified with dilute hydrochloric acid to pH=2, then allowed to cool. The resulting solid was collected on a filter, washed with water and air-dried to afford the title compound as a white solid (M+l 290). (US5824683, JOC 1946, 798).
[0189] Ethyl 2-(4'-bromobiphenyl-4-yl)acetate. To a solution of 2-(4'-bromobiphenyl-4- yl)acetic acid (2.5 g) in anhydrous ethanol (40 ml) was added concentrated sulfuric acid (1 ml). The mixture was refluxed for 2 h and the reaction was then concentrated. Water was added to the residue. The reaction was then neutralized with sodium bicarbonate and the mixture extracted with DCM (x3). The organic layers were combined, dried over sodium sulfate, and concentrated. The residue was purified by silica gel chromatography (eluted with 5%
EtOAc/Hexanes) to afford the title compound as an off-white solid (M+l 318).
[0190] Ethyl 2-(4'-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)biphenyl-4-yl)acetate (INT- 1). A mixture of ethyl 2-(4'-bromobiphenyl-4-yl)acetate (1.39 g, 4.39 mmol),
bis(pinacolato)diboron (1.67 g, 6.58 mmol), PdCl2(dppf)CH2Cl2 (0.359 g, 0.439 mmol) and potassium acetate (1.29 g, 13.2 mmol) in dioxane (50 ml) was heated in a sealed tube at 120°C for 18h. The cooled reaction mixture was filtered through celite and concentrated. The residue was purified by silica gel chromatography (eluted with 10-30% EtOAc/Hexanes) to afford the title compound as a light yellow oil (M+l 336.9).
Intermediate 2
Methyl 2-((lr,4r)-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)cvclohexyl)acetate

[0191] Methyl 2-(4-(4-hydroxyphenyl)cyclohexylidene)acetate. Sodium hydride (60% suspension in mineral oil, 1.2 g, 30 mmol) was added to a solution of 4-(4- hydroxyphenyl)cyclohexanone (5.7 g, 30 mmol) in anhydrous THF (100 mL) at 0 °C under a nitrogen atmosphere. In a separate flask, trimethyl phosphonoacetate (5.83 mL, 36 mmol) was added to a suspension of sodium hydride (1.8 g, 45 mmol) in anhydrous THF (100 mL) at 0 °C under a nitrogen atmosphere. After 30 min., the two mixtures were allowed to warm to rt and were stirred for 30 min. The solution containing the ketone was added to the phosphonate solution via cannula. The reaction mixture was stirred overnight at rt. Water (150 mL) was added, the organic layer was separated, and aqueous phase was extracted with EtOAc (2 x 120 mL). The combined organic layers were washed with brine, dried (MgS04), filtered, and concentrated in vacuo to provide the crude product. Purification by silica gel chromatography (10-20% EtOAc/Hexanes) yielded the title compound as a white solid. :H NMR (DMSO-d6) δ 9.12 (s, 1H), 7.01 (d, J = 8.4 Hz, 2H), 6.66 (d, J = 8.4 Hz, 2H), 5.70 (s, 1H), 3.82 (m, 1H), 3.61 (s, 3H), 2.70 (m, 1H), 2.33 (m, 2H), 2.03 (m, 1H), 1.92 (m, 2H), 1.48 (m,2H); (M+l 247.2).
[0192] Methyl 2-((lr,4r)-4-(4-hydroxyphenyl)cyclohexyl)acetate. A mixture of methyl 2-(4- (4-hydroxyphenyl)cyclohexylidene)acetate (5.72 g, 23.2 mmol) and 10% Pd/C (1.0 g) in EtOAc (150 mL) was stirred overnight at rt under a hydrogen atmosphere. The reaction mixture was filtered through celite and concentrated in vacuo to provide a white solid. :H NMR (DMSO-d6) δ 9.08 (s, ¾), 6.99 (d, J = 8.5 Hz, 2H), 6.65 (d, J = 8.5 Hz, 2H), 3.60 (s, 3H), 2.33 (dt, J = 3.0, 12.1 Hz, 1H), 2.23 (d, J = 6.9 Hz, 2H), 1.75 (m, 5H), 1.38 (m, 2H), 1.11 (m,2H); (M+l 249.2).
[0193] Methyl 2-((lr,4r)-4-(4-(trifluoromethylsulfonyloxy)phenyl)cvclohexyl)acetate. To a solution of methyl 2-((lr,4r)-4-(4-hydroxyphenyl)cyclohexyl)acetate (5.32 g, 21.4 mmol) and
triethylamine (4.5 mL, 32 mmol) in DCM (100 mL) was slowly added trifluoromethanesulfonic anhydride (4.5 ml, 27 mmol) under a nitrogen atmosphere. The reaction mixture was warmed to rt, stirred for 2 h, and then poured into water (50 mL). The layers were separated, and the aqueous phase was extracted with DCM (3X60 ml). The combined organic layers were washed with saturated aqueous NaHCC>3 and brine, then dried (MgS04), filtered, and concentrated in vacuo to provide the crude title compound as an off-white solid (M+l 381).
[0194] Methyl 2-((lr,4r)-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- vDphenvDcyclohexyDacetate (INT-2). A 250 mL round-bottomed flask was charged with methyl 2-((lr,4r)-4-(4-(trifluoromethylsulfonyloxy)phenyl)cyclohexyl)acetate (4.57 g, 10.7 mmol), KOAc (3.15 g, 32.1 mmol), bis(pinacolato)diboron (3.0 g, 11.8 mmol), PdCl2(dppf) (440 mg, 0.54 mmol), and dppf(297 mg, 0.54 mmol). The flask was consecutively filled with nitrogen and evacuated three times. Anhydrous dioxane (100 mL) was added and the reaction mixture was stirred overnight at 80°C. The cooled reaction mixture was diluted with EtOAc (300 mL), washed with brine, dried (MgS04), filtered, and concentrated in vacuo to provide a brown oil. Purification by flash chromatography (silica gel, 10-20% EtOA/hexane) provided the title compound as a white solid (M+l 359.3).
Intermediate 3
(R)-l-(2-chlorophenyl)ethyl 4-bromo-l -methyl- lH-pyrazol-5-ylcarbamate (INT-3a) and (R)-l- phenylethyl 4-bromo-l-methyl-lH-pyrazol-5-ylcarbamate (INT-3b)

[0195] To a stirred solution of phosgene (20% solution in toluene, 9.0 mL, 17.1 mmol), and molecular sieves (4A, 3.0 g) in anhydrous dichloromethane (-50 mL) at 0 °C under Ar was added anhydrous pyridine (5.0 mL, 62 mmol) dropwise, followed by 3-amino-4-bromo-2- methylpyrazole (1.0 g, 5.7 mmol) in dichloromethane (10 mL). This mixture was first stirred at
rt for 20 min, then at reflux for 40 min. The reaction mixture was then cooled to rt and (R) -2- chloro-a-methyl benzyl alcohol (3.0 g, 19.1 mmol) was added. The newly formed reaction mixture was refluxed for lh and then cooled to rt. The mixture was filtered and the filtrate was diluted with water (50 ml), and extracted with dichloromethane (3x 75 mL). The combined organic phases were washed with brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel chromatography to give (R)-l-(2-chlorophenyl)ethyl 4- bromo- 1 -methyl- lH-pyrazol-5-ylcarbamate (INT-3a) (M+l 357.8).
[0196] (R)-l-phenylethyl 4-bromo-l-methyl-lH-pyrazol-5-ylcarbamate (INT-3b) was prepared following analogous procedures (M+l 324, 326).
Intermediate 4
4-(4-Bromophenyl)-l-methyl-lH-pyrazol-5-amine

[0197] To 2-(4-bromophenyl)acetonitrile (20 g, 102 mmol) dissolved in EtOH (90 mL) was added l,l-dimethoxy-N,N-dimethylmethanamine (20.03 mL, 150 mmol) and stirred at ambient temperature for 48 hours. The mixture was concentrated to 50% volume, the suspension was filtered and the collected solid washed with diethyl ether (50 mL) to provide 2-(4-bromophenyl)- 3-(dimethylamino)acrylonitrile (M+H) 251.5.
[0198] Methyl hydrazine (27.4 mL, 520 mmol) was added to 2-(4-bromophenyl)-3- (dimethylamino)acrylonitrile (8.7 g, 34.6 mmol) at -20°C. The mixture was stirred at ambient temperature for 144 hours. The mixture was poured into water (50 mL) and stirred for 10 minutes. The precipitate was collected by filtration. The solid was purified by crystallization from a 1:1 mixture of ethanol:water (250 mL) to provide the title compound (M+H) 253.6.
Intermediate 5
l-Methyl-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)-lH-pyrazol-5-amine

[0199] To 4-(4-bromophenyl)-l-methyl-lH-pyrazol-5-amine (2.84 g, 11.26 mmol) dissolved in 1,4-dioxane (75 mL) was added potassium acetate (5.53 g, 56.3 mmol), 4,4,4',4',5,5,5',5'- octamethyl-2,2'-bi(l,3,2-dioxaborolane) (3.15 g, 12.39 mmol), tricyclohexylphosphine (0.758 g, 2.70 mmol) and palladium(0)bis(dibenzylideneacetone) (0.648 g, 1.126 mmol). The mixture was heated to reflux for 16 hours. The cooled reaction mixture was filtered through celite, washed with DCM and the filtrate concentrated in vacuo. The residue was purified by flash
chromatography (TBME to 2% MeOH in TBME) to provide the title compound (M+H) 300.3.
Intermediate 6
(R)-l-Phenylethyl l-methyl-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)-lH- -5-ylcarbamate

[0200] To l-methyl-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)-lH-pyrazol- 5-amine (INT-5, 1.052 g, 3.52 mmol) and pyridine (3.13 mL, 38.7 mmol) in DCM (50 mL) was added diphosgene (0.636 mL, 5.27 mmol) at 0°C and warmed to ambient temperature for 10 minutes. (R)-l-Phenylethanol (1.273 mL, 10.55 mmol) was added at 0°C. The mixture was stirred at ambient temperature for 16 hours. The mixture was poured into water (100 mL) and extracted with DCM (100 mL). The chlorinated layer was dried over magnesium sulfate, filtered and concentrated in vacuo. The residue was purified by flash chromatography (isohexane to 50% ethyl acetate in isohexane) to provide the title compound (M+H) 448.1.
Intermediate 7
3-(4-Bromophenyl)oxetane-3-carboxylic acid

[0201] (3-(4-Bromophenyl)oxetan-3-yl)methanol (250 mg, 1.028 mmol) was dissolved in acetonitrile (5 mL). 2,2,6, 6-Tetramethyl-l-piperidine 1-oxyl (TEMPO) (22.50 mg, 0.144 mmol) was added. A solution of sodium chlorite (372 mg, 4.11 mmol) and sodium hypochlorite (0.051 mL, 0.823 mmol) in water (2 mL) was added. The mixture was stirred at ambient temperature for 16 hours. The reaction was treated with 2M NaOH to pHIO, 10% sodium thiosulfate (2 mL) was added and the mixture partitioned between ethyl acetate and water. The aqueous phase was then acidified with citric acid and extracted with ethyl acetate. The organic phase was then dried over magnesium sulfate, filtered and concentrated in vacuo, to provide the title compound.
Intermediate 8
(R)- l-(2-Chlorophenyl)ethyl (4-(4-(tert-butoxycarbonylamino)phenyl)- 1-methyl- lH-pyrazol-5- yPcarbamate

[0202] A mixture of (R)-l-(2-chlorophenyl)ethyl-4-bromo-l-methyl-lH-pyrazol-5- ylcarbamate (INT-3a, 716 mg, 2.0 mmol), 4-(tert-butoxycarbonylamino)phenylboronic acid (711 mg, 3.0mmol), PdC^dppf'DCM (245 mg, 0.3 mmol), and sodium bicarbonate (340 mg, 4.0mmol) was degassed and flushed with argon. Dioxane (12 mL) and water (4 mL) were added and the mixture was heated at 90 °C with stirring for 16 h. The cooled mixture was filtered
through celite, and the filter cake was rinsed with ethyl acetate. The combined filtrate was washed with water and brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by silica gel chromatography to give the title compound (M+l 471.2).
Intermediate 9
4-(4-Chlorophenyl)-l-methyl-lH-pyrazol-5-amine

[0203] 4-(4-Chlorophenyl)-l-methyl-lH-pyrazol-5-amine was prepared from 4-bromo-l- methyl-lH-pyrazol-5-amine and (4-chlorophenyl)boronic acid following analogous procedures as described in Intermediate 8, using dichloro [Ι, bis(di-tert-butylphosphino)]ferrocene palladium (II) as catalyst, potassium phosphate as base, and dioxane/water as solvent, at 120 °C in a sealed reaction vessel, with a reaction time of 1 h.
Intermediate 10
Ethyl l-(4'-(5-amino-l-methyl-lH-pyrazol-4-yl)-ri, -biphenyll-4-yl)cvclopropanecarboxylate

[0204] Ethyl 1 - (4'-(5 -amino- 1 -methyl- 1 H-pyrazol-4-yl) - [ 1 , 1 '-biphenyl] -4- yl)cyclopropanecarboxylate was prepared from 4-(4-chlorophenyl)-l -methyl- lH-pyrazol-5- amine (INT-9) and ethyl l-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl)cyclopropanecarboxylate following the procedure in Intermediate 9, with a reaction temperature of 100 °C.
Intermediate 11
(R)-l-(2-Chlorophenyl)ethyl (4-(4-bromophenyl)-l-methyl-lH-pyrazol-5-yl)carbamate

[0205] A solution of (R)-l-(2-chlorophenyl)ethyl (4-(4-(tert-butoxycarbonylamino)phenyl)- 1 -methyl- lH-pyrazol-5-yl)carbamate (INT-8) in TFA/DCM (1 :4, 20 ml) was stirred at rt for 3 h. The reaction mixture was concentrated, then aqueous sodium bicarbonate solution was added to the residue, and the mixture was extracted with ethyl acetate. The combined organic phase was dried over sodium sulfate, filtered and concentrated to give (R)-l-(2-chlorophenyl)ethyl (4-(4- aminophenyl)-l-methyl-lH-pyrazol-5-yl)carbamate (M+l 370.9).
[0206] A stirred mixture of (R)-l-(2-chlorophenyl)ethyl (4-(4-aminophenyl)-l -methyl- 1H- pyrazol-5-yl)carbamate (528 mg, 1.42 mmol) and copper(II) bromide (350 mg, 1.57 mmol) in anhydrous acetonitrile was treated dropwise with t-butyl nitrite (425 uL, 3.57 mmol) at rt dropwise. After 2h the reaction mixture was concentrated in vacuo. Water was added to the residue and the mixture was extracted with ethyl acetate. The combined organic phase was dried over sodium sulfate, filtered and concentrated. The crude residue was purified by silica gel chromatography to provide the title compound (M+l 434.0).
Intermediate 12
Methyl 5-bromo-l-methyl-2,3-dihydro-lH-indene-l-carboxylate

(INT-12)
[0207] A stirred solution of methyl 5-bromo-2,3-dihydro-lH-indene-l-carboxylate (255 mg,
1.0 mmol) in anhydrous THF was treated with lithium bis(trimethylsilyl)amide (1.0 M in THF, 1.25 mL) dropwise at -78 °C. The reaction mixture was stirred at -78 °C for 45 min and then treated with methyl iodide (188 uL, 3.0 mmol) dropwise. The reaction mixture was allowed to warm to rt over 3 h, then was partitioned between ethyl acetate and water. The combined organic layers were dried over sodium sulfate, filtered and concentrated. The residue was purified by silica gel chromatography (ethyl acetate/hexanes eluent) to provide the title compound (M+l 269.0).
Intermediate 13
(R)-l-Phenylethyl (l-methyl-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)-lH- -5-yl)carbamate

[0208] A mixture of (R)-l-phenylethyl (4-(4-bromophenyl)-l-methyl-lH-pyrazol-5- yl)carbamate (prepared following analogous procedures described in Intermediate INT-11, 530 mg, 1.32 mmol), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(l,3,2-dioxaborolane) (673 mg, 2.65 mmol), dichloro l,l'-bis(diphenylphosphino)ferrocene palladium (II) dichloromethane complex (217 mg, 0.265 mmol ) and potassium acetate (390 mg, 3.98 mmol) was degassed and flushed with argon. Dioxane (10 mL) was added and the reaction mixture was heated at 100 °C with stirring for 3h. The cooled reaction mixture was filtered through a small celite plug eluted with ethyl acetate. The filtrate was then washed with water and brine sequentially. The combined organic phase was dried over sodium sulfate, filtered and concentrated. The residue was purified by silica gel chromatography (ethyl acetate/hexanes eluent) to provide the title compound (M+l 448.2).
Intermediate 14
4-Bromo-l-methyl-lH-l,2,3-triazole-5-carbaldehvde
9

(INT-14)
[0209] Bromine (2.2 mL, 43.4 mmol) was added cautiously to a solution of 1H-1,2,3- triazole (2 g, 29.0 mmol) in water (10 mL) at 40°C. The resulting mixture was stirred for 2 hrs at 40°C, then allowed to cool and stood overnight at room temperature. The precipitate was isolated by filtration, washed with water (2x10 mL), and dried in the vacuum oven to give 4,5- dibromo-lH-l,2,3-triazole as a light brown solid. (M+H 225.6; LCMS method 2).
[0210] Iodomethane (1.819 mL, 29.1 mmol) was added cautiously to a suspension of 4,5- dibromo-lH-l,2,3-triazole (4.4 g, 19.40 mmol) and potassium carbonate (5.36 g, 38.8 mmol) in THF (20 mL) at room temperature. The resulting mixture was stirred overnight. The reaction was poured into water (50 mL) and extracted with ethyl acetate (3x100 mL). The combined organics were washed with brine (50 mL), dried over sodium sulphate, filtered and concentrated in vacuo. Purification was by silica gel chromatography (eluted with a 0-50% ethyl acetate/isohexane) to give 4,5-dibromo-l-methyl-lH-l,2,3-triazole as a white solid (M+H 239.7; LCMS method 2).
[0211] Butyl lithium 1.6M in hexanes (3.89 mL, 6.23 mmol) was added dropwise over 5min to a solution of 4,5-dibromo-l-methyl-lH-l,2,3-triazole (1 g, 4.15 mmol) in THF (20 mL) at - 75 °C, maintaining the temperature <-70°C. The resulting mixture was then stirred for 1 hr at - 78°C. DMF (1.607 mL, 20.76 mmol) was then added and the mixture stirred for 30 min before being allowed to warm to room temperature. The reaction was quenched with 1M HCl (10 mL) and extracted with ether (3x50 mL). The combined organics were washed with brine (20 mL), dried over sodium sulphate, filtered and concentrated in vacuo. Purification was by silica gel chromatography (eluted with 0-50% ethyl acetate/isohexane) to give 4-bromo-l -methyl- 1H- l,2,3-triazole-5-carbaldehyde as a white solid (M+H30 207.9; LCMS method 2).
Intermediate 15
4-(4-Chlorophenyl)-l-methyl-lH-l,2,3-triazole-5-carboxylic acid

[0212] 4-Bromo-l-methyl-lH-l,2,3-triazole-5-carbaldehyde (INT-14, 250 mg, 1.316 mmol), 4-chlorophenylboronic acid (206 mg, 1.316 mmol) and l,l'-bis(di-tert- butylphosphino)ferrocene palladium dichloride (42.9 mg, 0.066 mmol) were charged to a microwave vial and the vial sealed. The vial was evacuated and refilled with nitrogen 5 times. Degassed dioxane (3.5 mL) and degassed potassium phosphate tribasic (1.761 mL, 2.237 mmol) (1.27M solution in water) was then added, the mixture stirred briefly then heated at 100°C for 1 hr in the microwave. The reaction was partitioned between DCM (20 mL) and water (lOmL). The layers were separated and the aqueous extracted with DCM (2x 20 mL). The combined organics were dried over sodium sulphate filtered and concentrated in vacuo. Purification was by silica gel chromatography (eluted with 0-50% ethyl acetate/isohexane) to give 4-(4- chlorophenyl)-l-methyl-lH-l,2,3-triazole-5-carbaldehyde as a white solid (M+H 221.9; LCMS method 2).
[0213] OXONE® (275 mg, 0.447 mmol) was added to a solution of 4-(4-chlorophenyl)-l- methyl-lH-l,2,3-triazole-5-carbaldehyde (90 mg, 0.406 mmol) in DMF (5 mL) and the resulting suspension was stirred at room temperature overnight. HC1 1M (10 mL) was added and the resulting mixture was extracted with ethyl acetate (3x10 mL). The combined organic solutions were washed with brine (2x10 mL), dried over sodium sulphate, filtered and concentrated in vacuo to give 4-(4-Chlorophenyl)-l-methyl-lH-l,2,3-triazole-5-carboxylic acid as a white solid (M+H 238.0; LCMS method 2).
Intermediate 16
(R)-l-Phenylethyl 4-(4-chlorophenyl)-l-methyl-lH-l,2,3-triazol-5-ylcarbamate

(INT-16)
[0214] To a suspension of 4-(4-chlorophenyl)-l-methyl-lH-l,2,3-triazole-5-carboxylic acid (INT-15, 111 mg, 0.467 mmol) in dry toluene (6 mL) was added 3 A molecular sieves, and then diphenyl phosphorazidate (0.100 mL, 0.467 mmol), triethylamine (0.065 mL, 0.467 mmol), and (R)-l-phenylethanol (0.169 mL, 1.401 mmol). The reagents were heated at 80°C for 3hrs. The reaction was filtered and the filtrate was concentrated in vacuo. Purification by silica gel chromatography (eluted with 0-50% ethylacetate/isohexane) gave the title compound as a white solid (M+H 357.4; LCMS method 2).
Intermediate 17
(R)-l-Phenylethyl (4-(5-bromopyridin-2-yl)-l-methyl-lH-pyrazol-5-yl)carbamate

O
X
CI^ I
Et3N, DMAP/DCM

[0215] A mixture of 2-(5-bromopyridin-2-yl)acetonitrile (2.085 g, 10.58 mmol), triethyl orthoformate (3.13 g, 21.2 mmol) and acetic anhydride (0.5 ml, 5.3 mmol) was stirred at 125 °C for 2h. Additional portions of triethyl orthoformate (3.13 g, 21.2 mmol) and acetic anhydride (0.5 ml, 5.3 mmol) were added, followed by stirring at 125 °C for 16h. The cooled reaction mixture was concentrated, then the residue was diluted with DCM (250 ml) and washed with brine. The organic layer was then dried over MgSC , filtered and concentrated. Purification by
silica gel chromatography gave 2-(5-bromopyridin-2-yl)-3-ethoxyacrylonitrile (M+l 253.1).
[0216] Methylhydrazine (0.38 ml, 7.16 mmol) was added to a solution of 2-(5- bromopyridin-2-yl)-3-ethoxyacrylonitrile (1.51 g, 5.97 mmol) in EtOH (20 ml),. The mixture was refluxed for 2h and then concentrated. The residue was purified by silica gel
chromatography (30-50% EtOAc/hexanes eluant) to give 4-(5-bromopyridin-2-yl)-l-methyl-lH- pyrazol-5 -amine. ]H NMR 400 MHz (DMSO-d6) δ 8.52 (d, 1H, J=2.4 Hz), 7.86 (dd, lHm J=1.6, 8.8 Hz), 7.77 (s, 1H), 7.51 (d, 1H, J=8.4 Jz), 6.43 (br, 2H), 3.58(s, 3H); (M+l 253.1).
[0217] A mixture of 4-(5-bromopyridin-2-yl)-l-methyl-lH-pyrazol-5-amine ( 253 mg, 1.0 mmol) and triethylamine ( 2.8 ml, 20 mmol) in DCM (30 ml) was cooled to 0 °C with stirring, then phosgene (5.26 ml, 1.9 M solution in toluene) was slowly added. The resulting mixture was stirred for lh at 0 °C. To this mixture was added a solution of (R)-l-phenylethanol (611 mg, 5.0mmol) in DCM (2.0 ml). Stirring was continued for 1 h at rt. 4- Dimethylaminopyridine (100 mg) was added to the reaction mixture, then the reaction vessel was sealed and the temperature was increased to 45 °C. After lh additional, a second portion of 4- dimethylaminopyridine (100 mg) was added, and the resulting mixture was stirred at 45 °C for 16h. The cooled reaction mixture was concentrated, then the residue was purified by reverse phase HPLC to give the title compound. ]H NMR 400 MHz (CDC13) δ 8.85 (s, 1H), 8.49 (d, 1H, J=2.4 Hz), 7.68 (m, 2H), 7.29-7.22 (m, 6H), 5.78 (q, 1H, J=2.8 Hz), 3.73 (s, 3H), 1.54 (d, 3H, J=6.8Hz); (M+ 1 401.1).
Intermediate 18
(R)-Ethyl 2-chloro-5-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)benzoate

(INT-18)
[0218] (R)-Ethyl 2-chloro-5-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4- yl)benzoate was prepared following analogous procedures described in Intermediate 9.
Intermediate 19
Methyl 6-(4-hydroxyphenyl)spiror2.51octane-l-carboxylate

[0219] 4-(4-(Benzyloxy)phenyl)cyclohexanone. Benzyl bromide (3.0 ml, 25 mmol) was added to a stirred mixture of 4-(4-hydroxyphenyl)cyclohexanone (3.96 g, 20.8 mmol), potassium carbonate (5.74 g, 41.6 mmol), and acetone (40 ml),. The mixture was then refluxed for 16h. After cooling to room temperature, the reaction mixture was concentrated, then the residue was diluted with water ( 60 ml) and extracted with DCM (3x120 ml). The combined extracts were then washed with brine (50 ml), dried over MgS04> filtered and concentrated. The residue was purified by silica gel chromatography (20-40% EtOAc/hexanes eluant) to afford the title compound (M+l 281.2).
[0220] Methyl 2-(4-(4-(benzyloxy)phenyl)cvclohexylidene)acetate. Sodium hydride (60% suspension in mineral oil, 440 mg, 11 mmol) was added to a solution of 4-(4- (benzyloxy)phenyl)cyclohexanone (2.8 g, 30 mmol) in anhydrous DMF (45 rriL) at 0 °C under a nitrogen atmosphere. The mixture was stirred for 15 minutes at 0 °C. A solution of trimethyl phosphonoacetate (2.0 g, 11 mmol) in DMF (4.0 ml) was added dropwise. The resulting mixture was stirred for 30 min at rt. Saturated NH4C1 solution was added, then the mixture was extracted with DCM (3x80 ml). The combined organic extracts were washed with brine, dried (MgS04), filtered, and concentrated in vacuo to provide the crude product. Purification by silica gel chromatography (10% EtOAc/hexanes eluant) yielded the title compound as a white solid. ]H NMR 400 MHz (CDC13) δ 7.47 (m, 4H), 7.36 (m, 1H), 7.15 (m, 2H), 6.94 (m, 2H), 5.70 (s,lH), 5.06 (s, 2H), 3.96 (m, 1H), 3.73 (s, 3H), 2.77 (m, 1H), 2.38 (m, 2H), 2.09 (m, 3H), 1.61 (m , 3H).
[0221] Methyl 6-(4-(benzyloxy)phenyl)spiror2.51octane-l-carboxylate. Sodium hydride (60% suspension in mineral oil, 619 mg, 15.4 mmol) was added to DMSO (40 ml) at rt, and the
mixture was stirred for 20 min at rt. Trimethylsulfoxonium iodide (3.40 g, 15.5 mmol) was added, and stirring was continued for 2h. A solution of methyl 2-(4-(4- (benzyloxy)phenyl)cyclohexylidene)acetate (1.74g, 5.16 mmol) was added. The resulting reaction mixture was stirred at rt for 2d, then water (100 ml) was added. The mixture was extracted with ether (4x60 ml), then the combined extracts were washed with brine, dried over MgS04, filtered and concentrated. The residue was purified by silica gel chromatography (0- 20% EtOAc/hexanes eluant) to yield the title compound. ]H NMR 400 MHz (CDC13)) δ 7.45 (m, 4H), 7.36 (m, 1H), 7.15 (m, 2H), 7.15 (m, 2H), 5.06 (s, 2H), 3.72 (s, 3H), 2.53 (m, 1H),2.02 (m, 1H), 1.85 (m, 4H), 1.62 (m, 2H), 1.25 (m, 2H), 1.03 (m , 1H), 0.96 (m, 2H); (M+l 351.2).
[0222] Methyl 6- (4-hydroxyphenyl) spiro Γ2.51 octane- 1 -carboxylate. A mixture of methyl 6- (4-(benzyloxy)phenyl)spiro[2.5]octane-l-carboxylate (1.183 g, 3.37 mmol ), 10% palladium on carbon (200 mg), and methanol (30 ml) was stirred at rt for 2h under a hydrogen atmosphere. The catalyst was removed by filtration. The filtrate was concentrated to afford the title compound (M+l 261.2).
Intermediate 20
Methyl 6-(4-(4,4,5,5-tetrametfayl-l,3,2-dioxaborolan-2-yl)phenyl)spiro[2.51octane-l-carboxylate

[0223] Methyl 6-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)spiro[2.5]octane-l- carboxylate (INT-20) was prepared following procedures described in Intermediate 2, steps 3 and 4.
Intermediate 21
2-Methoxy- 1 -phenylethanol

Intermediate 22
l-Phenylbut-2-yn-l-ol

[0225] Benzaldehyde (2.54 mL, 25 mmol) was added dropwise to prop-l-ynylmagnesium bromide 0.5M solution in THF (50 mL, 25.00 mmol) at -70°C under N2. The resulting mixture was stirred for 20min, allowed to warm to room temperature and stirred overnight. The reaction was quenched with saturated ammonium chloride solution (100 mL), and extracted with ether (3x100 mL). The combined organic solutions were washed with brine (100 mL), dried over magnesium sulphate, filtered and concentrated in vacuo . Purification was by silica gel chromatography (elute with 0-40% diethyl ether/isohexane) to afford the titled as a brown oil. ]H NMR (400MHz, CDC13) δ 7.56 (d, 2H), 7.45-7.30 (m, 3H), 5.45 (s, 1H), 2.19 (s, 1H), 1.93 (s, 3H).
Intermediate 23
3-Methoxy- 1 -phenylpropan- 1 -ol

[0226] 3 -Chloro-1 -phenylpropan- 1 -one (2 g, 11.86 mmol) and sodium iodide (1.778 g, 11.86 mmol) were combined in MeOH (35 mL) and refluxed overnight. The reaction was filtered and concentrated in vacuo. The residue was partitioned between ethyl acetate (100 mL) and brine (50 mL). The organic phase was separated, dried over sodium sulphate, filtered and
concentrated in vacuo. Purification by silica chromatography (eluted with 0-25% ethyl acetate/isohexane) gave 3-methoxy-l-phenylpropan-l-one as a colorless oil. ]H NMR (400MHz, CDCI3) δ 8.00 (dd, 2H), 7.59 (td, IH), 7.50 (td, 2H), 4.03 (t, 2H), 3.41 (s, 3H), 3.27 (t, 2H).
[0227] 3-Methoxy-l-phenylpropan-l-ol was obtained as a colorless oil from 3-methoxy-l- phenylpropan-l-one, following analogous procedures as described in Intermediate 21. ]H NMR (400MHz, CDCI3) δ 7.41-7.33 (m, 4H), 7.31-7.25 (m, IH), 4.93 (dd, IH), 3.66-3.54 (m, 2H), 3.39 (s, 3H), 2.97 (br. s, IH), 2.10-1.94 (m, 2H).
Intermediate 24
2-Ethoxy-l-phenylethanol (isomer mix)

[0228] A solution of benzaldehyde (2 g, 18.85 mmol) and (chloromethoxy)ethane (1.782 g, 18.85 mmol) in THF (25 mL) was added via syringe pump over 1 hr to a mixture of lithium (0.916 g, 132 mmol) and 4,4'-di-tert-butylbiphenyl (0.251 g, 0.942 mmol) in THF (50 mL) at 0°C. The reaction was stirred for a further 1 hr and cautiously quenched with water (25 mL). The reaction was stirred at room temperature until the lithium had been consumed. The reaction mixture was extracted with ethyl acetate (3x100 mL). The combined organic solutions were washed with brine (50 mL), filtered and concentrated in vacuo. Purification was by silica gel chromatography (eluted with 0-100% ethyl acetate/isohexane) to give the title compound as a colorless oil as a mixture of isomers. ]H NMR (400MHz, CDC13) δ 7.46-7.27 (m, 5H), 4.91 (dd, IH), 3.70-3.53 (m, 3H), 3.46 (t, IH), 2.82 (br, s, IH), 1.27 (t, 3H).
Intermediate 25
l-(Tetrahydro-2H-pyran-4-yl)ethanol

Intermediate 26
1 -(Tetrahydrofuran-3 -yPethanol

(INT-26)
[0230] Intermediate 26 was prepared in an analogous manner to l-(tetrahydro-2H-pyran-4- yl)ethanol (INT-25) replacing tetrahydro-2H-pyran-4-carbaldehyde with tetrahydrofuran-3- carbaldehyde.
Intermediate 27
Methyl l-(6-chloropyridin-3-yl)cvclopropanecarboxylate

[0231] 2-(6-Chloropyridin-3-yl)acetonitrile (lOg, 65.5mmol) and benzyltriethylammonium chloride (0.90g, 3.93mmol) were placed in a flask with l-bromo-2-chloroethane (16.3mL, 197mmoL). Sodium hydroxide solution (50% in water, 31.46g, 393 mmol) was added dropwise at 50°C and the reaction mixture was stirred at 50°C for 5 hours. Ethylene glycol (20mL) was
added and the reaction mixture was stirred at 100°C overnight. The reaction mixture was allowed to cool and acidified with 6M HC1. The product was extracted with ethyl acetate (6 x lOOmL). The organic phase was washed with water and brine, dried over MgS04, filtered and the solvent was removed in vacuo to give l-(6-chloropyridin-3-yl)cyclopropanecarboxylic acid (M+H 198.2).
[0232] l-(6-Chloropyridin-3-yl)cyclopropanecarboxylic acid (8g, 40.5mmol) was placed in a flask with dry MeOH (lOOmL). Concentrated sulfuric acid (2mL) was added and the reaction mixture was refluxed overnight. The solvent was removed in vacuo and the reaction mixture was partitioned between ethyl acetate and saturated NaHCC>3. The organic phase was washed with water and brine, dried over MgS04, filtered and the solvent was removed in vacuo to provide the title compound. (M+H 211.8). ]H NMR (400MHz, DMSO-i¾) δ 8.40 (1H, d), 7.85 (1H, d of d), 7.48 (1H, d), 3.59 (3H, s), 1.52 (2H, m), 1.30 (2H, m).
Intermediate 28
(4-Bromo-2-methyl-2H-pyrazol-3-yl)-carbamic acid 1,2,2,2-tetradeutero-l-phenyl-ethyl ester
(isomer mix)

[0233] To a solution of diphosgene (1.028 mL, 8.52 mmol) in DCM (20 mL) at 0°C was slowly added pyridine (5.05 mL, 62.5 mmol). The reaction mixture was stirred for 5 min at 0°C, warmed to room temperature, stirred for 30 min, and then (+/-)- l-phenylethan-l,2,2,2-D4-ol (1.434 g, 11.36 mmol) was added. The reaction mixture was stirred for 3 days at room temperature, diluted with water (20 mL) and DCM (100 mL). The layers were passed through a phase separator, and the organics concentrated in vacuo to provide the crude title compound as a brown oil. Purification by flash chromatography (silica gel, gradient ethyl acetate/hexane) provided the title compound as a white solid as a mixture of isomers (M+H 328.1).
Intermediate 29
Methyl l-(6-(4-hvdroxy-3-methoxyphenyl)pyridin-3-yl)cvclopropanecarboxylate

[0234] A mixture methyl l-(6-chloropyridin-3-yl)cyclopropanecarboxylate (INT-27, 332 mg, 1.567 mmol), 2-methoxy-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenol (400 mg, 1.599 mmol), dichloro [Ι, bis(di-tert-butylphosphino)]ferrocene palladium (II) (10.4 mg, 0.016 mmol), dioxane (3.7 mL), and water (0.74 mL) was purged with nitrogen for 30 min, then K3PO4 (1018 mg, 4.80 mmol), was added and heated at 120 °C in Biotage™ microwave for 60 min. Purification by flash chromatography (silica gel, gradient ethyl acetate/hexane) provided the crude title compound (M+l 300.2).
Intermediate 30
Methyl l-(6-(3-methoxy-4-(trifluoromethylsulfonyloxy)phenyl)pyridin-3- vDcyclopropanecarboxylate

[0235] To a mixture of methyl l-(6-(4-hydroxy-3-methoxyphenyl)pyridin-3- yl)cyclopropanecarboxylate (INT-29, 299 mg, 0.999 mol), pyridine (0.081 mL, 0.999 mol), and DCM (10 mL), was added trifluoromethanesulfonic anhydride 1M in DCM (0.999 mL, 0.999 mmol), The reaction mixture was stirred at room temperature for 3 days, additional
trifluoromethanesulfonic anhydride 1M in DCM (0.999 mL, 0.999 mmol) was added at 24 hrs
and 48 hrs. The reaction mixture was concentrated in vacuo to provide the crude title compound as a brown solid. Purification by flash chromatography (silica gel, gradient ethyl acetate/hexane) provided the title compound (M+l 432.2).
Intermediate 31
Methyl l-(6-(3-methoxy-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)pyridin-3- vDcyclopropanecarboxylate

[0236] In a Q-Tube a mixture of ([l,l'-bis(diphenylphosphino)ferrocene]
dichloropalladium(II) complex with dichloromethane (12.21 mg, 0.017 mmol), dioxane (0.5 mL), triethylamine (0.070 mL, 0.501 mmol), and 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(l,3,2- dioxaborolane) (72.5 mg, 0.285 mmol) and methyl l-(6-(3-methoxy-4- (trifluoromethylsulfonyloxy)phenyl)pyridin-3-yl)cyclopropanecarboxylate (INT-30, 72 mg, 0.167 mmol) was heated at 120 °C for 48 hrs then allowed to cool to room temperature further ([l,l'-bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with dichloromethane (48.8 mg, 0.067 mmol) added and heated at 120 °C for 4 days. Purification by flash
chromatography (silica gel, gradient ethyl acetate/hexane) gave the title compound as a white solid.
Intermediate 32
l-acetyl-4-(4-bromophenyl)piperidine-4-carboxylic acid

[0237] To 4-(4-bromophenyl)-l-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (500 mg,
1.301 mmol), in MeOH (10 mL) was added concentrated sulfuric acid (0.104 mL, 1.952 mmol) and stirred at room temperature for 72 hours. Concentrated sulfuric acid (0.050 mL, 0.938 mmol) was added and the reaction stirred at reflux for 36 hours. The cooled mixture was concentrated in vacuo, diluted with water (5 mL) and treated with 2M NaOH to pHIO and extracted with ethyl acetate. The organic layer was separated, dried over magnesium sulfate, filtered and concentrated in vacuo to give methyl 4-(4-bromophenyl)piperidine-4-carboxylate (M+H) 299.9.
[0238] To methyl 4-(4-bromophenyl)piperidine-4-carboxylate (85 mg, 0.285 mmol) in DCM (2 mL) was added pyridine (30 μΐ, 0.371 mmol) and acetyl chloride (50 μΐ, 0.703 mmol). The mixture was stirred at ambient temperature for 3 hours. 1M Hydrochloric acid (5 mL) was added and the chlorinated layer separated. The aqueous phase was extracted further with DCM, and the combined chlorinated layers were dried over magnesium sulfate, filtered and concentrated in vacuo to give methyl l-acetyl-4-(4-bromophenyl)piperidine-4-carboxylate (M+H) 342.0.
[0239] To methyl l-acetyl-4-(4-bromophenyl)piperidine-4-carboxylate (85 mg, 0.250 mmol) in THF (1 mL), water (0.5 mL) and MeOH (0.5 mL) was added lithium hydroxide monohydrate (52.4 mg, 1.249 mmol). The mixture was stirred at ambient temperature for 5 hours then treated with 1M Hydrochloric acid to pH 1-2 precipitating a white solid. The solid was collected by filtration washing with water to provide the title compound (M+H) 326.0.
Intermediate 33
4-(4-Bromopheny - 1 -(methylsulfonyl)piperidine-4-carboxylic acid

[0240] To methyl 4-(4-bromophenyl)piperidine-4-carboxylate (Intermediate 32, step 1) (85 mg, 0.285 mmol) in DCM (2 mL) was added pyridine (30 μΐ, 0.371 mmol) and methanesulfonyl chloride (30 μΐ, 1.35 mmol). The mixture was stirred at ambient temperature for 3 hours. Further methanesulfonyl chloride (30 μΐ, 1.35 mmol) was added and the mixture stirred at ambient temperature for 16 hours. Further methanesulfonyl chloride (50 μΐ, 0.642 mmol) and pyridine (50 μΐ, 0.618 mmol) were added and the mixture stirred for 16 hours. 1M Hydrochloric acid (5
mL) was added and the chlorinated layer separated. The aqueous phase was extracted further with DCM, and the combined chlorinated layers were dried over magnesium sulfate, filtered and concentrated in vacuo to give methyl 4-(4-bromophenyl)-l-(methylsulfonyl)piperidine-4- carboxylate (M+H) 377.9.
[0241] To methyl 4-(4-bromophenyl)-l-(methylsulfonyl)piperidine-4-carboxylate (98 mg, 0.260 mmol) in THF (1 mL), water (1 mL) and MeOH (1 mL) was added lithium hydroxide monohydrate (54.6 mg, 1.302 mmol). The mixture was stirred at ambient temperature for 16 hours then treated with 1M hydrochloric acid to pH 1-2 precipitating a white solid. The solid was collected by filtration washing with water to provide the title compound (M+H) 362.4.
Example la
2-(4'-(5-((l-(2-Chlorophenyl)ethoxy)carbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl-4- vDacetic acid

1a
[0242] A mixture of ethyl 2-(4'-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)biphenyl-4- yl)acetate (INT-1, 0.15 g, 0.41 mmol), 4-bromo-l -methyl- lH-pyrazol-5 -amine (87 mg, 0.49 mmol), Pd(PPh3)4 (47 mg, 0.041 mmol), and aqueous sodium carbonate (2M solution, 0.82 ml) in ethanol (1.6 ml) and toluene (8 ml) was heated in a sealed tube at 110°C for 4h. Water was added to the cooled reaction mixture, and the mixture was extracted with ethyl acetate (x3). The organic layers were combined, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel chromatography (eluted with 2% MeOH/DCM) to give ethyl 2-(4'-(5- amino- 1 -methyl- 1 H-pyrazol-4-yl)biphenyl-4-yl)acetate (M+ 1 336.1).
[0243] To a stirred solution of the above obtained mixture (300 mg), and anhydrous pyridine (0.71 g) in anhydrous DCM (10 ml) was added molecular sieves (1.2 g). Phosgene solution
(20% solution in toluene, 1.41 ml) was then added. This mixture was first stirred at room temperature for 20 min, and then at reflux for 40 min. The reaction mixture was then cooled to room temperature and a solution of +/- 2-chloro-a-methyl benzyl alcohol (282 mg) in DCM (5.0 ml) was added. The mixture was then refluxed for one hour. After cooling to room temperature, the reaction mixture was diluted with water (50 ml), and extracted with DCM (X3). The combined organic phases were dried over sodium sulfate, filtered, and evaporated. The residue was purified by HPLC (Ci8 column, eluted with 10-70% ACN/H20 with 0.035% TFA) to give ethyl 2-(4'-(5-((l-(2-chlorophenyl)ethoxy)carbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl- 4-yl)acetate (M+l 518.1).
[0244] To a stirred solution of ethyl 2-(4'-(5-((l-(2-chlorophenyl)ethoxy)carbonylamino)-l- methyl-lH-pyrazol-4-yl)biphenyl-4-yl)acetate (5) (47 mg) in 10:1 MeOH-H20 solution (10 ml) was added LiOH H20 (38 mg). The mixture was stirred at room temperature for 2h and then the solvent was removed under vacuum. Water was added to the residue, and the mixture was acidified with IN HC1 and then extracted with DCM (x3). The organic layers were combined, dried over sodium sulfate, filtered, and concentrated to give 2-(4'-(5-((l-(2- chlorophenyl)ethoxy)carbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl-4-yl)acetic acid racemate (la) as a white solid.
[0245] (R)-2-(4'-(5-(((l-(2-chlorophenyl)ethoxy)carbonyl)amino)-l-methyl-lH-pyrazol-4- yl)-[l,l'-biphenyl]-4-yl)acetic acid (lb) was prepared following similar procedures. HPLC-MS calculated for C27H25C1N304 (M+H) 490.2, found 490.2; RT 1.84 min. (LCMS method 1).

[0246] The following compounds were prepared following the procedures in Example 1, substituting appropriate reagents where required.

Structure
MS (m/z)
2-5 HPLC-MS calculated for C28H28N3O4
(M+H) 470.2, found 470.7; RT 1.14 min
(LCMS method 2)
2-6 HPLC-MS calculated for C24H26N3O4
(M+H) 420.2, found 420.6; RT 1.07 min
(LCMS method 2)
Example 3-1
2-((lr,4r)-4-(4-(5-((((R)-l-(2-Chlorophenyl)ethoxy)carbonyl)amino)-l-methyl-lH-pyrazol-4- yl)phenyl)cyclohexyl)acetic acid

[0247] Methyl 2-(( lr,4r)-4-(4-( 5 -(((R)- 1 -( 2-chlorophenyl)ethoxy)carbonylamino)- 1 -methyl- lH-pyrazol-4-yl)phenyl)cvclohexyl)acetate. A mixture of methyl 2-((lr,4r)-4-(4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)cyclohexyl)acetate (INT-2) (157 mg, 0.44 mmol), (R)-l-(2-chlorophenyl)ethyl 4-bromo-l-methyl-lH-pyrazol-5-ylcarbamate (INT-3a, 105 mg,
0.29 mmol), PdCl2(dppf) (24.5 mg, 0.03 mmol), NaHC03 ( 98 mg, 1.2 mmol), dioxane ( 4.0 mL), and water ( 1.5 mL) was heated at 100°C under a nitrogen atmosphere for 3 h. The mixture was diluted with water (15 mL) and extracted with EtOAc (3x30 mL). The combined extracts were washed with brine, dried over MgS04, and concentrated. The residue was purified by silica gel chromatography (20-50% EtOAc/Hexanes) to afford the title compound (M+l 510.3).
[0248] 2-((lr,4r)-4-(4-(5-(((R)-l-(2-Chlorophenyl)ethoxy)carbonylamino)-l-methyl-lH- pyrazol-4-yl)phenyl)cvclohexyl)acetic acid (3-1). A mixture of methyl 2-((lR,4r)-4-(4-(5-(((R)- l-(2-chlorophenyl)ethoxy)carbonylamino)-l-methyl-lH-pyrazol-4-yl)phenyl)cyclohexyl)acetate (73 mg, 0.145 mmol)), dioxane (5 ml), and aqueous lithium hydroxide (0.44 ml 2.0 M aq solution, 0.88 mmol) was stirred at 35°C for 16h. The reaction mixture was then acidified with 2N HC1, and the crude reaction mixture was purified directly by reverse-phase HPLC to afford the title compound.
[0249] Example 3-2 was prepared following analogous procedures as described in Example
3-1.

(R)-3-(4'-(l-methyl-5-(((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)-riJ'-biphenyll-4- -3-carboxylic acid

[0250] (R)-l-Phenylethyl l-methyl-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl)-lH-pyrazol-5-ylcarbamate (INT-6, 100 mg, 0.224 mmol), 3-(4- bromophenyl)oxetane-3-carboxylic acid (INT-7, 57.5 mg, 0.224 mmol), potassium phosphate tribasic (142 mg, 0.671 mmol) and [l,l'-bis(di-½ri-butylphosphino)ferrocene]
dichloropalladium(II) (7.28 mg, 0.011 mmol), were suspended in dioxane (1 mL) and water (0.25 mL) and degassed and flushed with nitrogen. The mixture was sealed and heated at 100°C for 30 minutes by microwave irradiation. The reaction mixture was treated with 10% aqueous citric acid to pH 5-6 and filtered through a pad of celite. The filtrate was partitioned between ethyl acetate and water. The aqueous phase was extracted further with ethyl acetate, and the combined organic layers were dried over magnesium sulfate, filtered and concentrated in vacuo. The residue was purified by preparative HPLC (C18 column eluted with 20-50% acetonitrile in water with 0.1% diethylamine, pH9) to give the title compound. HPLC-MS (M+H) 498.3; RT 1.08 min.
[0251] The following compounds were prepared following the procedures in Example 4-1, substituting appropriate reagents where required.

Example 5
(R)- 1 -phenylethyl ( 1 -methyl-4-( 4'-( 2-( methylsulfonamido)-2-oxoethyl)- [1,1 '-biphenyll-4-yl)- lH-pyrazol-5-yl)carbamate

(2-4)
[0252] Carbonyldiimidazole (17.1 mg, 0.11 mmol) was added to a solution of (R)-2-(4'-(l- methyl-5-(((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)-[l,l'-biphenyl]-4-yl)acetic acid (Example 2-4, 40 mg. 0.088 mmol) in DCM ( 3.0 ml). The resulting solution was stirred for 30 min at rt, then triethylamine (30 ul, 0.176 mmol) and methanesulfonamide (16.7 mg, 0.176 mmol) were consecutively added. The mixture was stirred at rt for 16h, then was concentrated. The residue was dissolved in DMSO and purified by reverse phase HPLC to provide the title compound.

Example 6a
(R)-l-(4'-(l-Methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4-

[0254] (R)-4-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)phenyl trifluoromethanesulfonate. (R)-l-phenylethyl 4-(4-hydroxyphenyl)-l -methyl- lH-pyrazol-5- ylcarbamate (130 mg, 0.385 mmol) was dissolved in anhydrous dichloromethane (2 mL) followed by addition of 4-(dimethylamino)pyridine (94 mg, 0.77 mmol) and
trifluoromethanesulfonic anhydride (163 mg, 0.58 mmol). The resulting solution was stirred at room temperature for 3 h. Water was added, then the mixture was extracted with
dichloromethane (3X). The combined organic layers were dried over sodium sulfate, filtered and evaporated. The residue was purified by silica gel chromatography (10% EtOAc/Hexanes to 100%EtOAc eluant) to yield the title compound (M+l 470).
[0255] (R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl- 4-yl)cyclopropanecarboxylic acid (6a). (R)-4-(l-methyl-5-((l-phenylethoxy)carbonylamino)- lH-pyrazol-4-yl)phenyl trifluoromethanesulfonate (60 mg, 0.13 mmol), l-(4- boronophenyl)cyclopropanecarboxylic acid (34 mg, 0.165 mmol), NaHCC>3 (33 mg, 0.39 mmol) and Pd(dppf)Cl2 (11 mg, 0.0135 mmol) were taken up in a mixture of dioxane (1 mL) and water (1 mL). The resulting mixture was heated at 90°C for 3 hours. The reaction was cooled and water was added, then the mixture was extracted with EtOAc (3X). The combined organic layers were dried over Na2SC>4, filtered and evaporated. The residue was purified by mass-triggered reverse-phase HPLC (Waters Micromass ZQ system, C18 column, Mobile phase A: H20 (0.05% TFA), Mobile phase B :ACN (0.035%TFA), flow rate 100 ml/min, 4 min gradient 10% to 90% B)to yield the title compound.(R)-l-(4'-(l-Methyl-5-((l-phenylethoxy)carbonylamino)-lH- pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid can also be prepared as follows:

[0256] (R)-Ethyl l-(4'-(l-methyl-5-(((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)- Γ 1 , 1 '-biphenyll -4-yl)cyclopropanecarboxylate. Pyridine (0.591 ml, 7.30 mmol) was added to a mixture of diphosgene (0.160 ml, 1.328 mmol) and molecular sieves (4A, 2g) in DCM (15 mL) under nitrogen at 0°C. The resulting mixture was stirred briefly and a solution of ethyl l-(4'-(5- amino- 1 -methyl- 1 H-pyrazol-4-yl)- [1,1 '-biphenyl] -4-yl)cyclopropanecarboxylate (INT- 10, 300 mg, 0.664 mmol) in DCM (5 mL) was added. The reaction was stirred for lOmin, refluxed for 40min then cooled to room temperature. (/?)-l-Phenylethanol (243 mg, 1.992 mmol) was then added and the reaction refluxed for 30min. Further (R)-l-phenylethanol (41mg, 0.33mmol) was added and the reaction was refluxed for a further 30min. The reaction was quenched with water (10 mL), diluted with DCM (20 mL) and the layers separated. The aqueous was extracted with DCM (20 mL) and the combined organics dried over sodium sulphate, filtered and concentrated in vacuo to give a brown gum. Purification by flash column chromatography eluting with a gradient of 0-50% ethyl acetate in iso-hexane afforded the title compound. [M+H]+ 510.3.
[0257] (R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)-ri,r- biphenyll-4-yl)cvclopropanecarboxylic acid (6a). A solution of lithium hydroxide monohydrate (69.2 mg, 1.648 mmol) in water (1 mL) was added to a solution of (R)-ethyl l-(4'-(l-methyl-5- (((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)-[l,l'-biphenyl]-4- yl)cyclopropanecarboxylate (210 mg, 0.412 mmol) in THF (2 mL) and the resulting mixture stirred vigorously at room temperature overnight. The reaction was then heated at 50°C for 90mins after which time MeOH (lmL) added and the mixture was stirred at room temperature for 2 days. The reaction was concentrated in vacuo to -lmL and carefully acidified with 1M
HC1, the resulting suspension was stirred vigorously for 30min. The precipitate was collected by filtration, washed with water (3x2mL), sucked dry and dried in a vacuum oven at 40°C to afford the title compound. HPLC-MS calculated for C29H28N3O4 (M+H) 482.2, found 482.1; RT 2.17 min.482.3 (LCMS Method 1). Melting point (Tm, onset) of 184 °C as determined by differential scanning calorimetry (DSC).
[0258] The racemate compound (6 racemate) was prepared from the racemate reactants following similar procedures. HPLC-MS calculated for C29H28N3O4 (M+H) 482.2, found 482.2; RT 1.24 min. (LCMS Method 2).

Example 6b
(R)-l-(4'-(l-Methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- vDcyclopropanecarboxylic acid potassium salt
[0259] To (R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylic acid (500 mg) was added KOH powder (58.3 mg) and a suitable solvent such as ethanol (10 mL). The resulting mixture was agitated (e.g., by shaking and/or sonication or by other suitable methods) until the solution became clear, followed by continued shaking for an additional two hours. Subsequently, the solvent was evaporated and the resulting solid was dried at 40 °C in a vacuum over for 12 hours. The dried product was scratched to powder to give the amorphous potassium salt.
[0260] To the amorphous potassium salt was added a suitable solvent such as ethyl acetate, 3 -methyl- 1-butanol or 4-methyl-2-pentanone (7 mL), resulting in a slurry which was stirred at room temperature for approximately 72 hours. The solvent was then removed, and the crystalline salts were collected and dried in a vacuum oven at 40 °C for 24 hours.
[0261] Crystalline formation of (R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)- lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid potassium salt was confirmed by X- Ray Powder diffraction (XRPD) pattern (Figure 1), and exhibits the following thermal parameters: melting point (Tm, onset) of 150 °C as determined by differential scanning calorimetry (DSC) at a scanning rate of 10 ° C/min (Figure 2A); decomposition point of 175° C, and a weight loss on drying of 3.4%, as determined by thermogravimmetric (TGA) analysis (Figure 2B). In some embodiments, crystalline (R)-l-(4'-(l-methyl-5-((l- phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid potassium salt is characterized as having an XRPD diffraction pattern substantially the same as Figure 1; a DSC thermogram substantially the same as Figure 2A, a TGA thermogram substantially the same as Figure 2A; or combinations thereof.
[0262] In situ salt formation. To_(R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)- lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid (5 mg) was added a KOH/H2O solution (0.25 M, 50 \lL). The mixture was stirred and heated at 55 °C to dissolve visible solids; subsequently, 50 \lL of a microemulsion preconcentrate (e.g., percentage by weight: 16.7% ethanol, 8.3% propylene glycol, 16.9% LABRAFIL®M2125CS and 58.1% CREMOPHOR EL®) was added to the mixture, which was stirred to give a clear solution with a pH of -8.5.
Example 7
[0263] The following compounds were prepared following the procedures in Example 6, substituting appropriate reagents where required.


Example 8
(R)-3-(4'-(5-(((l-(2-chlorophenyl)ethoxy)carbonyl)amino)-l-methyl-lH-pyrazol-4-yl)-ri, - biphenyll-4-yl)propanoic acid

[0264] (R)-l-(2-chlorophenyl)ethyl 4-(4-bromophenyl)-l-methyl-lH-pyrazol-5-ylcarbamate (INT-11, 44 mg, 0.10 mmol), methyl 3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl)propanoate (31 mg, 0.15 mmol), dichloro l,l '-bis(diphenylphosphino)ferrocene palladium (II) dichloromethane complex (18 mg, 0.022 mmol ) and sodium bicarbonate (18 mg, 0.21 mmol) were combined, degassed and flushed with Ar. Dioxane (1.5 mL) and water (0.5 mL) were added and the mixture was stirred and heated at 90 °C for 16h. The cooled mixture was filtered through a small celite plug eluted with ethyl acetate. The filtrate was then washed with water, and brine sequentially. The combined organic phases were dried over sodium sulfate, filtered and concentrated. The residue was purified by preparative thin layer
chromatography to provide (R)-3-(4'-(5-(((l-(2-chlorophenyl)ethoxy)carbonyl)amino)-l-methyl- lH-pyrazol-4-yl)-[l,l'-biphenyl]-4-yl)propanoic acid methyl ester (M+l 518.2).
[0265] A mixture of (R)-3-(4'-(5-((l-(2-chlorophenyl)ethoxy)carbonylamino)-l-methyl-lH- pyrazol-4-yl)biphenyl-4-yl)propanoic acid methyl ester (48mg, 0.09 mmol),, THF (6 mL), MeOH (2 mL), water (2 mL), and lithium hydroxide monohydrate (40 mg, 0.95 mmol) was stirred at rt for 30 min. The reaction mixture was then concentrated in vacuo. The crude residue was then treated with acetic acid (1 mL), and partitioned between ethyl acetate and water. The combined organic phase was washed with water (1 x 10 mL), dried over sodium sulfate, filtered and concentrated to provide the title compound.
1H NMR 400 MHz and/or
Structure
Cpd # MS (m/z) (LCMS method 1)
8
HPLC-MS calculated for C28H27CIN3O4 (M+H) 504.2, found 504.1; RT 2.17 min.
Example 9
[0266] The following compounds were prepared following the procedures in Example 8, substituting appropriate reagents where required.


- Ill -

Example 10-1
l-Methyl-5-(4-(l-methyl-5-((((R)-l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)phenyl)-

[0267] (R)-l-Phenylethyl (l-methyl-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl)-lH-pyrazol-5-yl)carbamate (INT- 13, 81 mg, 0.20 mmol), methyl 5-bromo-l-methyl-
2,3-dihydro-lH-indene-l-carboxylate (INT-12, 81 mg, 0.30 mmol), 1,1'- bis(diphenylphosphino)- ferrocene-palladium(II)dichloride dichloromethane complex (33 mg, 0.04 mmol) and cesium bicarbonate (130 mg, 0.40 mmol) were combined, degassed and flushed with argon. Dioxane (3.0 mL) and water (1.0 mL) were added and the mixture was heated with stirring at 90 °C for 16h. The cooled reaction mixture was treated with 1 M hydrochloric acid to pH 3-4, then treated with ethyl acetate (30mL) and filtered through a small celite plug. The filtrate was then partitioned between ethyl acetate and water. The collected aqueous phase was extracted further with ethyl acetate, and the combined organic layers were dried over sodium sulfate, filtered and concentrated. The residue was purified by preparative thin layer chromatography, which gave methyl l-methyl-5-(4-(l-methyl-5-((((R)-l- phenylethoxy)carbonyl)amino)- lH-pyrazol-4-yl)phenyl)-2,3-dihydro- lH-indene- 1-carboxylate as an isomeric mixture (M+l 510.2).
[0268] To the solution of methyl l-methyl-5-(4-(l-methyl-5-((((R)-l- phenylethoxy)carbonyl)amino)- lH-pyrazol-4-yl)phenyl)-2,3-dihydro- lH-indene- 1-carboxylate in THF/MeOH/water (3:1:1, 10 ml) was added lithium hydroxide (100 mg, 4.2 mmol). The reaction mixture was stirred at rt for 16h, then was concentrated under reduced pressure. The residue was partitioned between ether and water. The aqueous layer was treated with acetic acid to pH 4-5, then was extracted with ethyl acetate. The ethyl acetate extract was concentrated to give l-methyl-5-(4-(l-methyl-5-((((R)-l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4- yl)phenyl)-2,3-dihydro-lH-indene-l-carboxylic acid as a mixture of isomers. The isomeric mixture was subjected to chiral HPLC conditions (Column: 21x25mm Whelk O-l; Solvents: Hexane/MeOH/EtOH (7:2:1) with 1% HO Ac; Flow Rate: 1 mL/min; Temperature: ambient; Run Time: 25 mins) to give: (i) Isomer A (Retention time: 17.08 mins); and (ii) Isomer B
(Retention time: 22.24 mins).
[0269] Example 10-2 was prepared following the procedures in Example 10-1, substituting appropriate reagents where required.
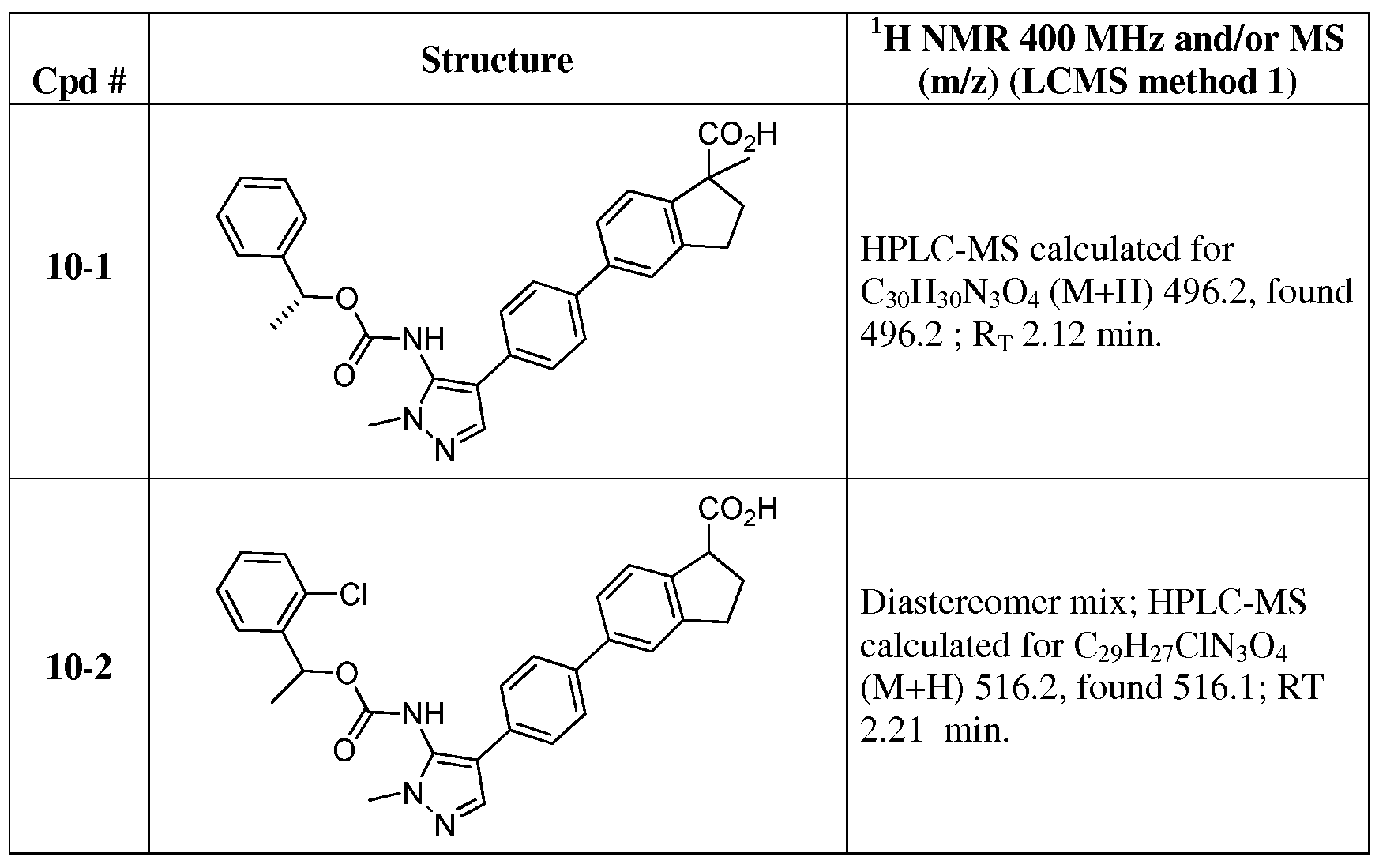
Example 11
(R)-l-(4'-(l-methyl-5-(((l-phenylethoxy)carbonyl)amino)-lH-l,2,3-triazol-4-yl)-ri, - biphenyll-4-yl)cvclopropanecarboxylic acid

[0270] (R)-l-Phenylethyl 4-(4-chlorophenyl)-l-methyl-lH-l,2,3-triazol-5-ylcarbamate (INT-16, 120 mg, 0.336 mmol), l-(4-boronophenyl)cyclopropanecarboxylic acid (76 mg, 0.370 mmol) and l,l'-bis(di-tert-butylphosphino)ferrocene palladium dichloride (10.96 mg, 0.017 mmol) were charged to a microwave vial and the vial sealed. The vial was evacuated and refilled with nitrogen 5 times. Degassed dioxane (0.9 mL) and degassed potassium phosphate tribasic (0.450 mL, 0.572 mmol) (1.27M solution in water) was then added, the mixture stirred briefly then heated at 100°C for 2 hr in the microwave. The reaction was acidified with acetic acid and partitioned between ethyl acetate (10 mL) and water (10 mL). The layers were separated and the aqueous extracted with ethyl acetate (2x10 mL). The combined organics were washed with
brine (10 mL), dried over sodium sulphate filtered and concentrated in vacuo. Purification was by silica gel chromatography (eluted with 50-100% ethyl acetate/isohexane to afford the title compound as a tan solid.

Example 12
[0271] The following compounds were prepared following analogous procedures described in Example 6, substituting appropriate reagents where required.


Example 13-1
2-Acetamido-3-(4'-(l-methyl-5-((((R)-l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)-ri, - biphenyll-4-yl)propanoic acid

[0272] 2-Acetamido-3-(4'-(l-methyl-5-((((R)-l-phenylethoxy)carbonyl)amino)-lH-pyrazol- 4-yl)-[l,l'-biphenyl]-4-yl)propanoic acid was prepared following the procedures described in Example 8, step 1.
[0273] Example 13-2 was prepared following analogous procedures described in Example -1, substituting appropriate reagents where required.

Example 14
(R)-l-(4-(6-(l-methyl-5-(((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)pyridin-3- yl)phenyl)cyclopropanecarboxylic acid

[0274] A mixture of (R)-l-phenylethyl (4-(5-bromopyridin-2-yl)-l-methyl-lH-pyrazol-5-
yl)carbamate (INT- 17, 67 mg, 0.17 mmol), (4-(l-(methoxycarbonyl)cyclopropyl)phenyl) boronic acid (74.8 mg, 0.34 mmol), PdCl2(dppf) ( 12.4 mg, 0.017 mmol), NaHC03 (57.1 mg, 0.68 mmol), dioxane (4.0 ml), and water (1.0 ml) was heated with stirring in a sealed tube at 95 °C for 3h. After cooling to rt, the crude reaction mixture was filtered, and the filtrate was purified by reverse-phase HPLC to give (R)-methyl l-(4-(6-(l-methyl-5-(((l- phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)pyridin-3-yl)phenyl)cyclopropanecarboxylate. (M+l 497.3). ]H NMR 400 MHz (CD3OD) δ 8.78 (d, IH, J=2.0Hz), 8.38 (dd, IH, J=1.6, 8.4 Hz), 8.05 (s, IH), 7.82 (d, IH, J=8.4Hz), 7.67 (d, 2H, J=8.4 Hz), 7.55 (d, 2H, J=8.8Hz), 7.33 (m, 5H), 5.76 (q, IH, J=2.4 Hz), 3.80 (s, 3H), 3.64 (s, 3H), 1.65 (m, 2H), 1.60 (d, 3H, J=6.8 Hz), 1.28 (m, 2H).
[0275] A solution of (R)-methyl l-(4-(6-(l-methyl-5-(((l-phenylethoxy)carbonyl)amino)- lH-pyrazol-4-yl)pyridin-3-yl)phenyl)cyclopropanecarboxylate (52 mg, 0.105 mmol) in dioxane (4 ml) was treated with lithium hydroxide (2.0 M solution, 1.04 ml) and the mixture was stirred at 45 °C for 4h. The mixture was allowed to cool to rt, then 2N hydrochloric acid was added to pH 6.0. This crude mixture was then purified by reverse-phase HPLC to provide the title compound.

Example 15
(R)-l-(2'-(hydroxymethyl)-4'-(l-methyl-5-(((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4- yl)-rl,r-biphenyH-4-yl)cyclopropanecarboxylic acid

[0276] To a solution of (R)-ethyl 2-chloro-5-(l-methyl-5-((l-phenylethoxy)carbonylamino)- lH-pyrazol-4-yl)benzoate (INT- 18, 150 mg, 0.35 mmol) in 10 ml anhydrous DCM at 0 °C was slowly added DIBAL-H (1.75 ml of 1M solution in CH2CI2, 1.75 mmol). The resulting solution was then stirred at 0 °C for 1.5 h. Ice was added, the organic layer was separated, and the aqueous phase was extracted with EtOAc (3 X 150 ml). The combined organic layers were dried over Na2S04, filtered and evaporated. The residue was purified by silica gel flash
chromatography to give (R)-l-phenylethyl (4-(4-chloro-3-(hydroxymethyl)phenyl)-l-methyl- lH-pyrazol-5-yl)carbamate.
[0277] (R)-Methyl l-(2'-(hydroxymethyl)-4'-(l-methyl-5-(((l- phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)-[l,l'-biphenyl]-4-yl)cyclopropanecarboxylate was prepared following similar procedures described in Intermediate 10; and the corresponding carboxylic acid was prepared following similar procedures described in Example 6 (method 2), step 2.

(R)-l-(6-(4-(l-methyl-5-(((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)phenyl)pyridin-3- vDcyclopropanecarboxylic acid

[0278] (R)-methyl l-(6-(4-(l-methyl-5-(((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4- yl)phenyl)pyridin-3-yl)cyclopropanecarboxylate was prepared following analogous procedures as described in Example 13-1, with methyl l-(6-bromopyridin-3-yl)cyclopropanecarboxylate as reactant. Ester hydrolysis following similar procedures described in Example 6 (Method 2), step
2, gave the title compound.

Example 17
6-(4-(l-Methyl-5-((((R)-l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4- yl)phenyl)spiror2.51octane-l-carboxylic acid (isomer mix)

[0279] The title compound was prepared following similar procedures described in Example
3-1.

biphenyll-4-yl)cvclopropanecarboxylic acid
[0280] l-(4'-(5-(((l-Deutero-l-phenylethoxy)carbonyl)amino)-l-methyl-lH-pyrazol-4-yl)- [l,l'-biphenyl]-4-yl)cyclopropanecarboxylic acid was prepared as a racemic mixture (18-1) following the procedures in Example 6 substituting the appropriate reagents where required. The isomeric mixture was subjected to chiral SFC conditions (Column: Chiralcel OJ-H 250 x 10 mm, 5 μιη; Mobile phase: 50% methanol / 50% C02; Flow rate: 10 mL/min; Detection: UV @ 220 nm) to give: (i) Isomer A (SFC Retention Time: 2.78 mins), LCMS RT 4.02min; MS m/z 483.1 [M+H]+ (LCMS method 3); and (ii) Isomer B (SFC Retention Time: 4.34 min). LCMS RT 4.11min; MS m/z 483.3 [M+H]+ (LCMS method 3).
[0281] Examples 18-2 and 18-3 were prepared following the procedures in Example 18-1
substituting appropriate reagents where required.

Example 19
l-(4'-(5-((2-methoxy-l-phenylethoxy)carbonylamino)- l-methyl-lH-pyrazol-4-yl)biphenyl-4- vDcyclopropanecarboxylic acid

[0282] To diphosgene (0.250 mL, 2.075 mmol) in DCM was added pyridine (1.231 mL, 15.22 mmol) and stirred at ambient temperature into solution. Ethyl l-(4'-(5-amino-l-methyl- lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylate (INT-10, 500 mg, 1.383 mmol) was added, the mixture stirred at ambient temperature for 30 minutes. 2-Methoxy-l-phenylethanol (INT-21, 444 mg, 2.92 mmol) was added and the mixture was stirred at ambient temperature for 36 hours. The mixture was concentrated in vacuo and the residue purified by flash
chromatography (TBME to 5% MeOH in TBME) to give ethyl l-(4'-(5-(((2-methoxy-l- phenylethoxy)carbonyl)amino)- 1 -methyl- lH-pyrazol-4-yl)- [1,1 '-biphenyl]-4- yl)cyclopropanecarboxylate. (M+H) 540.6.
[0283] To ethyl l-(4'-(5-((2-methoxy-l-phenylethoxy)carbonylamino)-l-methyl-lH- pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylate (130 mg, 0.241 mmol) in THF (2 mL), water (2 mL) and MeOH (2 mL) was added lithium hydroxide monohydrate (101 mg, 2.409 mmol) and stirred at ambient temperature for 2 hours. The mixture was added to 2M HC1 (10 mL) and extracted with ethyl acetate, the organic layer was dried over magnesium sulfate, filtered and concentrated in vacuo to give the title compound as a mixture of enantiomers (19); (M+H) 512.3.
[0284] The enantiomeric mixture was subjected to chiral SFC conditions (Column:
Chiralpak AD-H, 250 x 10 mm, 5 μιη @ 35 °C; Mobile phase: 40% isopropanol / 60% C02; Flow rate: 10 mL/min; Detection: UV @ 220 nm) to give: (i) Isomer A (SFC Retention Time: 5.11 mins), LCMS RT 1.07 min; MS m/z 512.3 [M+H]; LCMS method 2; and (ii) Isomer B
(SFC Retention Time: 6.37 min), LCMS RT 1.05 min; MS m/z 512.4 [M+H]; LCMS method 2.

vDcvclopropanecarboxylic acid
[0285] Methyl l-(4'-(l-methyl-5-((l-phenylbut-2-ynyloxy)carbonylamino)-lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylate was prepared as a yellow foam following procedures described in Example 19, step 1, with l-phenylbut-2-yn-l-ol (INT-22) as reactant. Rt 1.31min; MS m/z 520.4 [M+H]+; Method 2.
[0286] Lithium hydroxide monohydrate (9.69 mg, 0.231 mmol) was added to a solution of methyl l-(4'-(l-methyl-5-((l-phenylbut-2-ynyloxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylate (30 mg, 0.058 mmol) in a mixture of THF (0.5 mL) and water (0.500 mL) and the resulting mixture stirred overnight. The reaction was acidified with acetic acid, diluted with water (5 mL) and extracted with ethyl acetate (3x10 mL). The combined organic solutions were washed with brine (5 mL), dried over sodium sulphate, filtered and concentrated in vacuo. The residue was taken up in DCM (2 mL) and MP-Isocyanate resin (100 mg, 3eq, 0.149mmol, 1.49 mmol/g) was added. The resulting mixture was stirred overnight. The resin was removed by filtration and washed with DCM (3x5 mL). The combined filtrate and washings were concentrated in vacuo. The residue was triturated with iso-hexane to give the title compound as an off white solid (isomeric mixture).

Example 21
l-(4'-(5-((3-Methoxy-l-phenylpropoxy)carbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl-4- yDcyclopropanecarboxylic acid

[0287] Ethyl l-(4'-(5-((3-methoxy-l-phenylpropoxy)carbonylamino)-l-methyl-lH-pyrazol- 4-yl)biphenyl-4-yl)cyclopropanecarboxylate was prepared as a yellow foam following the procedures described in Example 19 step 1, with 3-methoxy-l-phenylpropan-l-ol as reactant. (M+H 554.5).
[0288] Lithium hydroxide monohydrate (61.4 mg, 1.463 mmol) was added to a solution of ethyl l-(4'-(5-((3-methoxy-l-phenylpropoxy)carbonylamino)-l-methyl-lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylate (135 mg, 0.244 mmol) in a mixture of water (1.000 mL) and THF (1 mL) and the resulting mixture stirred at room temperature for 90 mins then heated to 50°C for 30 hrs. The reaction was acidified using 1M HC1 solution and the resulting mixture extracted with ethyl acetate (3x10 mL). The combined organic solutions were washed with brine (5 mL), dried over sodium sulphate, filtered and evaporated. The residue was dissolved in DCM (10 mL) and MP-isocyanate resin (200 mg 0.298 mmol, 1.49 mmol/g) was added. The resulting mixture was stirred overnight at room temperature. The resin was removed by filtration, washed with DCM (3x5 mL) and the combined washings and filtrate concentrated in vacuo. The residue was triturated with ether to give an enantiomeric mixture of l-(4'-(5-((3-methoxy-l-phenylpropoxy)carbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid as a white solid (21); (M+H 526.5).
[0289] The enantiomeric mixture was subjected to chiral SFC conditions (Column:
Chiralpak AD-H, 250 x 10 mm, 5 μιη @ 35 °C; Mobile phase: 40% Methanol / 60% C02; Flow rate: 10 mL/min; Detection: UV @ 220 nm) to give: (i) Isomer A (SFC Retention Time: 4.10 mins), LCMS Rt 5.07min; MS m/z 526.5 [M+H]+; (LCMS method 4); and (ii) Isomer B (SFC Retention Time: 5.10 min), LCMS Rt 5.07min; MS m/z 526.4 [M+H]+; (LCMS method 4).

l-(4'-(5-(((cvclopropyl(phenyl)methoxy)carbonyl)amino)-l-methyl-lH-pyrazol-4-yl)-rU'- biphenyll-4-yl)cvclopropanecarboxylic acid

[0290] Ethyl l-(4'-(5-((cyclopropyl(phenyl)methoxy)carbonylamino)-l-methyl-lH-pyrazol- 4-yl)biphenyl-4-yl)cyclopropanecarboxylate (isomer mix) was prepared as a yellow foam following the procedures described in Example 19 step 1, with (+)cyclopropyl(phenyl)methanol as reactant. (M+H 536.4).
[0291] Lithium hydroxide monohydrate (61.4 mg, 1.463 mmol) was added to a solution of ethyl l-(4'-(5-((cyclopropyl(phenyl)methoxy)carbonylamino)-l-methyl-lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylate (135 mg, 0.244 mmol) in a mixture of water (1.0 mL) and THF (1 mL) and the resulting mixture stirred at room temperature for 90 min. The reaction was heated to 50°C for 30 hrs. The reaction was acidified using 1M HC1 solution and the resulting mixture extracted with ethyl acetate (3x10 mL). The combined organic solutions were washed with brine (5 mL), dried over sodium sulphate, filtered and evaporated. The residue was triturated with ether to give l-(4'-(5-((cyclopropyl(phenyl)methoxy)carbonylamino)-l-methyl- lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid (isomer mix) as a white solid. (M+H 508.3).

Example 23
l-(4'-(5 -((2-Ethoxy- 1 -phenylethoxy)carbonylamino)- 1 -methyl- 1 H-pyrazol-4- yl)biphenyl-4- vDcyclopropanecarboxylic acid (isomer mix)

[0292] Ethyl 1 -(4'-(5-((2-ethoxy- 1 -phenylethoxy)carbonylamino)- 1 -methyl- lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylate (isomer mix) was prepared as a yellow foam following procedures described in Example 19 step 1, with 2-ethoxy-l-phenylethanol (INT-24) as reactant. (M+H 554.5)
[0293] A solution of lithium hydroxide monohydrate (57.6 mg, 1.373 mmol) in water (1.000 mL) was added to a solution of ethyl l-(4'-(5-((2-ethoxy-l-phenylethoxy)carbonylamino)-l- methyl-lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylate (95 mg, 0.172 mmol) in ethanol (1 mL) and the resulting mixture stirred at 50°C for 90 min and stood at room temperature for 2 weeks. The reaction was acidified with 1M HCl solution (3 mL) and concentrated in vacuo. Purification was by preparative HPLC (C18 column eluted with 0-100% acetonitrile in water with 0.1% TFA) to give the title compound as an off-white solid mixture of isomers (M+H 526.6).

(R)-l-(4'-(l-Methyl-5-((3-methylbutan-2-yloxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- vDcyclopropanecarboxylic acid
[0294] Step la) To a stirred solution of diphosgene (551μΕ, 4.57mmol) in anhydrous DCM (80 mL) at 0°C under nitrogen was added anhydrous pyridine (2.71mL, 33.5mmol) dropwise. The reaction mixture was allowed to return to room temperature. Ethyl l-(4'-(5-amino-l-methyl- lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylate (INT-10, 500mg, 1.38mmol) in DCM (30 mL) was then added. This mixture was stirred at room temperature for 30 minutes to form a stock solution.
[0295] Step lb) To a stirred solution of (R)-3-methylbutan-2-ol (73mg, 0.828mmol) in anhydrous dichlorome thane (1 mL) at room temperature under nitrogen was added a 10 mL aliquot of the previously prepared solution and the newly formed reaction mixture was stirred at room temperature for 2 hours. The solvent was removed in vacuo and the residue was purified by silica gel chromatography to provide the title compound. [M+H] 476.4.
[0296] A mixture of (R)-ethyl l-(4'-(l-methyl-5-((3-methylbutan-2-yloxy)carbonylamino)- lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylate (80mg, 0.169mmol), THF (2 mL), water (1 mL), and lithium hydroxide monohydrate (24.27mg, 1.014mmol) was stirred at 50 °C for 2 days. The reaction mixture was acidified with 1M HC1, diluted with water (lOmL), extracted with DCM (lOmL) and passed through a phase separator. Isohexane (5mL) was added and the solvent was removed in vacuo to give the title compound as a white solid. (M+H 448.1).
[0297] Example 24b was prepared following the procedures in Example 24a substituting appropriate reagents where required.

Structure
Cpd # MS (m/z) (LCMS method 2)
HPLC-MS calculated for
24b
o OH C26H30N3O4 (M+H) 448.2,
found 448.4; RT 1.17min.
Example 25
l-(4'-(5-((l-Cyclopropylethoxy)carbonylamino)-l-mem^
yDcyclopropanecarboxylic acid
[0298] Ethyl l-(4'-(5-((l-cyclopropylethoxy)carbonylamino)-l-methyl-lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylate (racemate) was prepared from 1-cyclopropylethanol following the procedure described in Example 24a, step 1. (M+H 474.3).
[0299] SFC chiral separation of ethyl l-(4'-(5-((l-cyclopropylethoxy)carbonylamino)-l- methyl-lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylate (isomer mix) was carried out under the following conditions: (Column: Chiralpak IC, 250 x 10 mm, 5μιη; mobile phase: A = 2-propanol + 0.1% diethylamine / B = C02; Flow rate: 10 mL / min; detection: UV @ 254 nm) to give: (i) enantiomer A: (SFC Retention Time: 9.83 mins); and (ii) enantiomer B: (SFC Retention Time: 10.29 min).
[0300] l-(4'-(5-((l-Cyclopropylethoxy)carbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl- 4-yl)cyclopropanecarboxylic acid as a racemic mixture (25) was obtained as a white solid from ethyl l-(4'-(5-((l-cyclopropylethoxy)carbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl-4-yl) cyclopropanecarboxylate (racemate) following the procedure described in Example 24a, step 2.
[0301] l-(4'-(5-((l-Cyclopropylethoxy)carbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl- 4-yl)cyclopropanecarboxylic acid (Isomer A) and l-(4'-(5-((l- Cyclopropylethoxy)carbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid (Isomer B) were obtained as a pink solid from the
corresponding ethyl l-(4'-(5-((l-cyclopropylethoxy)carbonylamino)-l-methyl-lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylate enantiomer A and enantiomer B, respectively, following similar procedures. For further purification, the product was dissolved in DCM (9mL)
and THF (lmL) and treated with MP-NCO (400 mg, 1.49 mmol/g) for 18 hours at room temperature to remove amine impurity. The resin was filtered off, isohexane (5 mL) was added and the solvent was removed in vacuo to give the title compound (Isomer A and Isomer B).

Example 26
l-(4'-(l-Methyl-5-((l-o-tolylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yDcyclopropanecarboxylic acid
[0302] Ethyl l-(4'-(l-methyl-5-((l-o-tolylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl- 4-yl)cyclopropanecarboxylate was prepared following the procedure used in Example 24a, step 1, with (+)-l-o-tolylethanol as reactant. (M+H 524.3).
[0303] l-(4'-(l-Methyl-5-((l-o-tolylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid was prepared from ethyl l-(4'-(l-methyl-5-((l-o- tolylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylate following the procedure used in Example 24a step 2. The product was further purified following the same procedures described in Example 25, to give a white solid as a mixture of isomers (26), (M+H 496.3).
[0304] SFC chiral separation of l-(4'-(l-methyl-5-((l-o-tolylethoxy)carbonylamino)-lH- pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid was carried out under the following conditions: (Column: Chiralcel OJ-H 250 x 10 mm, 5 μιη; mobile phase: 45% methanol / 55% C02; Flow rate: 10 ml/min; detection: UV @ 220 nm) to give: l-(4'-(l-methyl-5-((l-o- tolylethoxy)carbonylamino)- lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid (peak 1, Isomer A); SFC Retention Time: 4.04 mins. Rt 1.18 mins; MS m/z 496.4 [M+H]+; LCMS Method 2.
[0305] l-(4'-(l-methyl-5-((l-o-tolylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid (peak 2, Isomer B); SFC Retention Time: 5.89 min. Rt 1.19
mins; MS m/z 496.3 [M+H]+; LCMS Method 2.

yl)biphenyl-4-yl)cvclopropanecarboxylic acid
[0306] Ethyl l-(4'-(l-methyl-5-((l-(tetrahydro-2H-pyran-4-yl)ethoxy)carbonylamino)-lH- pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylate was prepared following the procedure described in Example 24a, step 1, with l-(tetrahydro-2H-pyran-4-yl)ethanol (INT-25) as reactant. (M+H 518.4).
[0307] l-(4'-(l-Methyl-5-((l-(tetrahydro-2H-pyran-4-yl)ethoxy)carbonylamino)-lH- pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid was prepared from ethyl l-(4'-(l- methyl-5-((l-(tetrahydro-2H-pyran-4-yl)ethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylate, following the procedure described in Example 24a, step 2. The product was further purified following the same procedures described in Example 25, to give a white solid as a mixture of isomers (27); (M+H 490.3).
[0308] SFC chiral separation of l-(4'-(l-methyl-5-((l-(tetrahydro-2H-pyran-4-yl)ethoxy) carbonylamino)-lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid was carried out under the following conditions: (Column: Phenomenex LUX C2 250 x 10 mm, 5 μιη; mobile phase: 40% methanol / 60% C02; flow rate: 10 mL/min; detection: UV @ 220 nm) to give: (i) Peak 1: l-(4'-(l-methyl-5-((l-(tetrahydro-2H-pyran-4-yl)ethoxy) carbonylamino)-lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylic acid Isomer A. SFC Retention Time: 8.75 min;
HPLC-MS calculated for C28H32N3O5 (M+H) 490.2, found 490.6; RT 1.06 min. LCMS Method 2; and (ii) Peak 2: l-(4'-(l-methyl-5-((l-(tetrahydro-2H-pyran-4-yl)ethoxy) carbonylamino)-lH- pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid Isomer B. SFC Retention Time: 11.42
min; HPLC-MS calculated for C28H32N3O5 (M+H) 490.2, found 490.4; RT 1.05 min. LCMS Method 2.

Example 28
l-(4'-(l-Methyl-5-((l-(tetrahvdrofuran-3-yl)ethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-
4-yl)cvclopropanecarboxylic acid
[0309] l-(4'-(l-Methyl-5-((l-(tetrahydrofuran-3-yl)ethoxy)carbonylamino)-lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylic acid as a mixture of isomers (28) was prepared following the procedures in Example 27, replacing l-(tetrahydro-2H-pyran-4-yl)ethanol
(INT25) with l-(tetrahydrofuran-3-yl)ethanol (INT-26) as reactant. (M+H 476.3).
[0310] SFC chiral separation of l-(4'-(l-methyl-5-((l-(tetrahydrofuran-3- yl)ethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylic acid (isomer mix) was carried out under the following conditions: (Column: Chiralcel OJ-H 250 x 10 mm, 5 μιη [2 columns coupled together]; mobile phase: 35% methanol / 65% C02; flow rate: 10 mL/min; detection: UV @ 220 nm) to give: (i) peak 1 (Isomer A; SFC Retention Time: 6.52 min; Rt 1.04 mins; MS m/z 476.3 [M+H]+; LCMS method 2); and (ii) peak 2 (Isomer B; SFC Retention Time: 8.64 mins; Rt 1.04 mins; MS m/z 476.5 [M+H]+; LCMS method 2); and (iii) peak 3 (Isomer C; SFC Retention Time: 11.58 min; Rt 1.03 mins; MS m/z 476.3 [M+H]+; LCMS method 2); and (iv) peak 4 (Isomer D; SFC Retention Time: 12.98 minRt 1.03 mins; MS m/z 476.4 [M+H]+; LCMS method 2).
1H NMR 400 MHz and/or
Structure
Cpd # MS (m/z) (LCMS method 2)
HPLC-MS calculated for
28
C27H30N3O5 (M+H) 476.2, °v0H found 476.3; RT 1.04 min.
Example 29- 1
l-(4'-(5-(Benzyloxycarbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl-4- vDcvclopropanecarboxylic acid
[0311] Ethyl l-(4'-(5-(benzyloxycarbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylate and methyl l-(4'-(5-(benzyloxycarbonylamino)-l-methyl-lH- pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylate (3: 1 mixture). To a stirred solution of diphosgene (501μΕ, 4.15mmol) in anhydrous DCM (80 mL) at 0°C under nitrogen was added anhydrous pyridine (2.71mL, 33.5mmol) dropwise. The reaction mixture was allowed to return to room temperature, and ethyl l-(4'-(5-amino-l-methyl-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylate (INT- 10, 800mg, 2.22mmol) and methyl l-(4'-(5-amino-l-methyl- lH-pyrazol-4-yl)biphenyl-4-yl)cyclopropanecarboxylate (200mg, 0.55mmol) in DCM (20 mL) were then added to form a stock solution, which was stirred at room temperature for 30 minutes.
[0312] To a stirred solution of phenylmethanol (90mg, 0.831mmol) in anhydrous dichloromethane (1 mL) at room temperature under nitrogen was added a 10 mL aliquot of the previously prepared stock solution, and the reaction mixture was stirred at room temperature for 2 hours. The solvent was removed in vacuo and the residue was purified by silica gel chromatography to provide the title compound mixture. Rt 1.26 mins (methyl ester - 25%); MS m/z 482.3 [M+H]+; (LCMS method 2); Rt 1.31 mins (ethyl ester - 75%); MS m/z 496.3
[M+H]+; (LCMS method 2).
[0313] l-(4'-(5-(Benzyloxycarbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid was prepared from a mixture of ethyl l-(4'-(5- (benzyloxycarbonylamino)- 1 -methyl- lH-pyrazol-4-yl)biphenyl-4-yl) cyclopropanecarboxylate and methyl l-(4'-(5-(benzyloxycarbonylamino)-l-methyl-lH-pyrazol-4-yl)biphenyl-4-
yl)cyclopropanecarboxylate (3 : 1 mixture) following the procedure used in Example 24a, step 2. (M+H 468.3).
[0314] Examples 29-2 through Examples 29-4 were prepared following the procedures in
Example 29- 1 substituting appropriate reagents where required.

(R)-l-(6-(4-(l-methyl-5-((3-methylbutan-2-yloxy)carbonylamino)-lH-pyrazol-4- yl)phenyl)pyridin-3-yl)cvclopropanecarboxylic acid hydrochloride
[0315] Methyl l-(6-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)pyridin-3- vDcyclopropanecarboxylate. Methyl l-(6-chloropyridin-3-yl)cyclopropanecarboxylate (INT-27, 500mg, 2.36mmol), l,4-bis(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzene (1.56mg, 4.72mmol) and l,l'-bis(di-tert-butylphosphino)ferrocene palladium dichloride (77mg,
0.118mmol) were placed in a flask with dioxane (lOmL) tribasic potassium phosphate (852mg, 4.02mmol), dissolved in water (2mL), was added and the reaction mixture was stirred at 100°C for 1 hour. The dioxane was removed in vacuo and the reaction mixture was partitioned between DCM and water. The organic phase was passed through a phase separator and the solvent was removed in vacuo. The residue was purified by silica gel chromatography (0-100% ethyl acetate in isohexane) to provide the title compound. (M+H 380.3). ]H NMR (400MHz, DMSO- ) δ 8.67 (1H, d), 8.11 (2H, d), 7.97 (1H, d), 7.87 (1H, d of d), 7.79 (2H, d), 3.60 (3H, s), 1.58 (2H, m), 1.31 (14H, m).
[0316] Methyl l-(6-(4-(5-amino-l-methyl-lH-pyrazol-4-yl)phenyl)pyridin-3- vDcyclopropanecarboxylate. Methyl l-(6-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl)pyridin-3-yl)cyclopropanecarboxylate (290mg, 0.765mmol), 4-bromo-l -methyl- 1H- pyrazol-5 -amine (148mg, 0.841mmol) and l,l'-bis(di-tert-butylphosphino)ferrocene palladium dichloride (24.9mg, 0.038mmol) were placed in a flask with dioxane (5mL). Tribasic potassium phosphate (276mg, 1.30mmol), dissolved in water (lmL), was added and the reaction mixture was stirred at 100°C for 1 hour. The dioxane was removed in vacuo and the reaction mixture was partitioned between DCM and water. The organic phase was passed through a phase separator and the solvent was removed in vacuo. The residue was purified by silica gel chromatography (0-100% ethyl acetate in isohexane) to provide the title compound. (M+H 349.3). ]H NMR (400MHz, DMSO- ) δ 8.61 (1H, d), 8.04 (2H, d), 7.90 (1H, d), 7.80 (1H, d of d), 7.57 (2H, d), 7.51 (1H, s), 5.48 (2H, s), 3.62 (3H, s), 3.60 (3H, s), 1.58 (2H, m), 1.31 (2H, m).
[0317] (R)-methyl l-(6-(4-(l-methyl-5-((3-methylbutan-2-yloxy)carbonylamino)-lH- pyrazol-4-yl)phenyl)pyridin-3-yl)cyclopropanecarboxylate was prepared from methyl l-(6-(4- (5-amino-l-methyl-lH-pyrazol-4-yl)phenyl)pyridin-3-yl)cyclopropanecarboxylate and (R)-3- methylbutan-2-ol following the procedure used in Example 24a, step 1. (M+H 463.5).
[0318] (R)-l-(6-(4-(l-methyl-5-((3-methylbutan-2-yloxy)carbonylamino)-lH-pyrazol-4- yl)phenyl)pyridin-3-yl)cyclopropanecarboxylic acid hydrochloride was prepared from (R)- methyl l-(6-(4-(l-methyl-5-((3-methylbutan-2-yloxy)carbonylamino)-lH-pyrazol-4- yl)phenyl)pyridin-3-yl)cyclopropanecarboxylate following the procedure used in Example 24a, step 2. The residue was purified by reverse phase chromatography (C18 column eluted with 0- 95% acetonitrile in water with 0.1% HC1) to provide the title compound. (M+H 449.3).
[0319] Examples 30-2 and 30-3 were prepared following the procedures in Example 30-1 substituting appropriate reagents where required.

Example 31
1 -(6- { 3-Methoxy-4- Γ 1 -methyl-5 -( 1 ,2,2,2-tetradeutro- 1 -phenyl-ethoxycarbonylamino)- 1 H- pyrazol-4-yll-phenyl|-pyridin-3-yl)-cvclopropanecarboxylic acid hydrochloride

[0320] 1 -(6- { 3-Methoxy-4- Γ 1 -methyl-5-( 1 ,2,2,2-tetradeutro- 1 -phenyl- ethoxycarbonylamino)-lH-pyrazol-4-yll-phenyl|-pyridin-3-yl)-cvclopropanecarboxylic acid methyl ester (isomer mix). A mixture of methyl l-(6-(3-methoxy-4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)phenyl)pyridin-3-yl)cyclopropanecarboxylate (INT-31, 36 mg, 0.049 mmol), (4-bromo-2-methyl-2H-pyrazol-3-yl)-carbamic acid 1,2,2,2-tetradeutro-l-phenyl-ethyl ester (INT-28, 16.17 mg, 0.049 mmol), dichloro [Ι, bis(di-tert-butylphosphino)]ferrocene palladium (II) (0.321 mg, 0.493 mmol), dioxane (1 mL), and water (0.2 mL) was purged nitrogen for 30 min, then K3PO4 (31.4 mg, 0.148 mmol), added and heated at 120 °C in Biotage™ microwave for 60 min. Purification by flash chromatography (silica gel, gradient ethyl acetate/hexane) provided the title compound as a mixture of isomers (M+l 531.4).
[0321] l-(6-{ 3-Methoxy-4-ri-methyl-5-(l,2,2,2-tetradeutro-l-phenyl- ethoxycarbonylamino)-lH-pyrazol-4-yll-phenyll-pyridin-3-yl)-cyclopropanecarboxylic acid hydrochloride (isomer mix). A mixture of l-(6-{3-methoxy-4-[l-methyl-5-(l,2,2,2-tetradeutro- 1 -phenyl-ethoxycarbonylamino)- 1 H-pyrazol-4-yl] -phenyl } -pyridin-3 -yl)- cyclopropanecarboxylic acid methyl ester (isomer mix) (4.1 mg, 7.73 μιηοΐ), MeOD-<¾ (0.75 mL), water (0.1 mL), and lithium hydroxide (1.62 mg, 0.039 mmol) was stirred at room temperature for 7 days. The reaction mixture was concentrated in vacuo, the residue was diluted with 2M NaOH (0.5 mL), and extracted with DCM (1.5 mL). The aqueous phase was acidified with 5M HC1, extracted with DCM (2x3 mL), and the organic layers were dried via a phase separator. The organic phase was concentrated in vacuo to give the title compound mixture of isomers as a yellow solid (M+l 517.7).

Example 32
(R)-l-Acetyl-4-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)piperidine-4-carboxylic acid
[0322] (R)-l-Phenylethyl l-methyl-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl)-lH-pyrazol-5-ylcarbamate (INT-6, 100 mg, 0.224 mmol), l-acetyl-4-(4- bromophenyl)piperidine-4-carboxylic acid (INT-32, 72.9 mg, 0.224 mmol), potassium phosphate tribasic (142 mg, 0.671 mmol) and [l,l'-bis(di-ieri- butylphosphino)ferrocene]dichloropalladium (II) (7.28 mg, 0.011 mmol), were suspended in dioxane (2 rriL) and water (0.5 iriL) and degassed and flushed with nitrogen. The mixture was sealed and heated at 100°C for 30 minutes by microwave irradiation. The reaction mixture was treated with 1M HC1 (2 rriL) and filtered through a pad of celite. The filtrate was partitioned between ethyl acetate and water. The aqueous phase was extracted further with ethyl acetate, and the combined organic layers were dried over magnesium sulfate, filtered and concentrated in vacuo. The residue was purified by preparative HPLC (C18 column eluted with 20-50% acetonitrile in water with 0.1% diethylamine, pH9) to provide the title compound (M+H) 567.4

Example 33
(R)-4-(4'-(l-methyl-5-(((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4-yl)-ri ,l'-biphenyll-4- vD- 1 -(methylsulfonyl)piperidine-4-carboxylic acid
[0323] (R)-l-phenylethyl l-methyl-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl)-lH-pyrazol-5-ylcarbamate (INT-6, 88 mg, 0.196 mmol), 4-(4-bromophenyl)-l- (methylsulfonyl)piperidine-4-carboxylic acid (INT-33, 71 mg, 0.196 mmol), potassium phosphate tribasic (125 mg, 0.588 mmol) and [l,l'-bis(di-½ri-butylphosphino)ferrocene] dichloropalladium(II) (6.4 mg, 0.098 mmol), were suspended in dioxane (2 mL) and water (0.5 mL) and degassed and flushed with nitrogen. The mixture was sealed and heated at 100°C for 30 minutes by microwave irradiation. The reaction mixture was treated with 1M HC1 and ethyl acetate then filtered through a pad of celite. The filtrate was partitioned between ethyl acetate and water. The aqueous phase was extracted further with ethyl acetate, and the combined organic layers were dried over magnesium sulfate, filtered and concentrated in vacuo. The residue was purified by preparative HPLC (C18 column eluted with 20-50% acetonitrile in water with 0.1% diethylamine, pH9) to provide the title compound (M+H) 603.4

Example 34
(R)-l-Phenylethyl 4-(4'-(l-(2-cvanoethylcarbamoyl)cvclopropyl)biphenyl-4-yl)-l-methyl-lH- pyrazol-5-ylcarbamate
[0324] To (R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4- yl)biphenyl-4-yl)cyclopropanecarboxylic acid (Example 6a) (500 mg, 1.038 mmol) in DMF (10 mL) was added N,N,N',N'-tetramethyl-0-(7-azabenzotriazol-l-yl)uronium hexafluorophosphate (HATU) (434 mg, 1.142 mmol), triethylamine (0.434 mL, 3.11 mmol) and aminopropanenitrile (0.085 mL, 1.090 mmol). The mixture was stirred at ambient temperature for 16 hours. The mixture was poured into water (100 mL) and extracted with ethyl acetate (2 x 100 mL), the combined organics were dried over magnesium sulfate, filtered and concentrated in vacuo to provide the title compound (M+H) 534.7.

[0325] The activity of a compound according to the present invention can be assessed by the following methods, infra.
[0326] LPA1, LPA2, and LPA3 FLIPR Assays in ChemK-1 Cells for Antagonist Selectivity (Method 1). ChemK-1 cells stably transfected with human LPA1 or human LPA3 expression vectors were purchased from Millipore. ChemK- 1 cells transfected with a human LPA2 expression vector were generated according to standard procedures. Cells were harvested using trypsin/EDTA, diluted in media to 0.6 x 106 cells/mL and 20 of cells were added to a 384 well FLIPR plate to give 12000 cells/well. Next day, medium is removed from cells and replaced with 15μί loading buffer (Molecular Devices Calcium No- Wash Assay Kit 4 (4 x stock) diluted to lx stock in assay buffer), and cells loaded for 45 min to 1 hour and incubated at 37 °C. For antagonist studies, 40 minutes into loading time, 5 μL· of test compound (solution in DMSO/ Assay Buffer) or buffer control was added online using a Hamamatsu FDSS 7000 to the 384 well cell plate, and incubated for an additional 40-45 min at 37 °C. 5 μL· of agonist cocktail (LPA at EC80 concentration) or buffer control was added online and the plate was read using the Hamamatsu FDSS/7000.
[0327] Running the assay in antagonist mode, the following IC50 values were obtained for Example 1 :
Table 1

[0328] LPA1 FLIPR Antagonist Assay in Chem-1 cells (Method 2). 15,000 LPAl-Cheml cells (Millipore) are plated in a volume of 25 uL of DMEM with 1% FBS, lx Non-Essential Amino Acids and 10 mM HEPES in a 384 well plate. Cells are incubated at 37 °C overnight and the next day 25 uL of FLIPR dye [Fluo-4 Direct FLIPR Dye (Invitrogen) reconstituted in FLIPR buffer (lx HBSS with 20mM Hepes)] with freshly added 5 mM probenecid (Sigma) and 0.1% fatty acid free Bovine Serum Albumen (Sigma). 500 mL of test compound is transferred into the assay plate for 10 min before 12.5 uL of agonist cocktail (500 nM LPA final concentration in FLIPR buffer + 0.1% fatty acid free BSA) is added and the signal is read on a FLIPR Tetra instrument (Molecular Devices) in a 384 well setting. Compound IC50S are determined by standard 4-parameter fit equations.
[0329] hLPAl GTPyS Binding Assay in Cheml Cells (Method 1). Chem-1 cells, overexpressing the LPAi receptor, were grown to 80-90 % confluency in 500 cm2 cell-culture trays at 37°C/5 C02. All subsequent steps were conducted at 4°C to avoid receptor degradation. The cell-culture medium was removed and ice cold buffer (10 ml per tray; lOmM HEPES, 0.9 % w/v NaCl, 0.2 % w/v EDTA, pH 7.4) was added to the cells, scraped from the trays into a 50 mL Corning tube and subsequently centrifuged at 250 x g for 5 min. The supernatant fraction was aspirated and 10 mL"1 500 cm2 tray of wash buffer (lOmM HEPES, lOmM EDTA, pH 7.4) was added to the pellet. This was homogenized using an electrical homogenizer ' Werker, ultra-turrax' (position 6, 4 x 5 s bursts) and subsequently centrifuged at 48,000 x g at 4°C (Beckman Avanti J-251 Ultracentrifuge) for 30 min. The supernatant was discarded and the pellet re-homogenized and centrifuged as described above, in wash buffer. The final pellet was suspended in ice cold assay buffer (lOmM HEPES, O. lmM EDTA, pH 7.4) at a concentration of 1-5 mg mL"1. Protein concentration was determined by the bicinchoninic acid assay based on the method of Smith (1985), using BSA as a standard and aliquots maintained at -80°C until required.
[0330] Test compound solutions for ten-point concentration response curves (10 - 0.0003μΜ) were prepared at lOOx concentration in 100% DMSO, and 2.5μΙ, added to a 96-well white OptiPlate. A concentration of LPA, corresponding to an ECso concentration, prepared in assay buffer (20mM HEPES, lOmM MgCl2, lOOmM NaCl and ImM EDTA) was added to each well containing test compound in a volume of 2.5μί. To each well, 200μΕ of assay buffer, with 0.1% BSA and 30μg mL"1 saponin, added fresh on the day of experimentation, containing 50μg mL"1 membranes, 3.7μΜ guanosine 5 '-diphosphate sodium salt (GDP) and 2.5mg mL"1 WGA PVT SPA beads was added to each well and incubated at room temperature with gentle agitation for 30min, allowing equilibrium to be reached. Following this, 50μL· of [35S]-Guanosine 5'-(γ- thio)triphosphate (GTPyS) at a concentration of 300pM was added to each well, and incubated at room temperature with gentle agitation for 40min. Following this the plates were centrifuged for 3min at 3,000rpm before being quantified using single photon counting on a TopCount™ microplate scintillation counter (Perkin Elmer, Beaconsfield, UK).
[0331] hLPAl GTPyS Binding Assay in Cheml Cells (Method 2). Homogenized membranes are prepared from LPAl-Cheml cells purchased from Millipore. Harvested LPA1- Cheml cells are resuspended in 20mM Hepes, pH 7.4 with lOmM EDTA and protease inhibitor tablet (Roche) at one tablet per 25ml of buffer. After homogenization, cell lysate is centrifuged
at 50,000g for 25min to pellet the cell membranes. The membrane is resuspended in 20mM Hepes, pH 7.4 + 0.1 mM EDTA + 15% glycerol + 1 tablet of protease inhibitor per 10ml buffer and stored at -80°C.
[0332] Solutions of test compounds ranging from 10 mM to 0.01 nM are prepared in DMSO. The desired amount of membrane prep is diluted with ice-cold assay buffer (20mM HEPES, pH7.4, lOOmM NaCl, lOmM MgCl2, 0.2% fatty acid free BSA, lOug/ml saponin, fresh add 2uM GDP) and vortexed well. 2 ul or less of compound is distributed into each well of a round-bottom 96-well polystyrene assay plate, followed by addition of 150 ul of diluted membranes (1-10 ug/well) and kept on ice until the addition of hot GTPgammaS. [35S]- GTPgammaS is diluted 1: 10000 (v/v) with cold assay buffer and 50 ul is added into each well. The reaction is carried out at room temperature for 2 hours before the membranes are harvested onto PerkinElmer Unifilter GF/B-96 filter plate using a Packard Filtermate Harvester. After several washes with wash buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2), the filter plate is dried at room temperature for 30 minutes. MicroScint-20 is added and the plate sealed for scintillation counting on TopCount (PerkinElmer). EC50 values are obtained by fitting the GTP [gamma-35S] binding curves (raw data) with the dose response curve-fitting tool of GraphPad Prism. IC50 values in the GTPyS binding assay are given in Examples above as indicated.
[0333] Human Lung Fibroblast (HLF) assay. HLFs (Promocell, Heildelberg, Germany) were plated in full growth medium (DMEM with 10% FBS and Na Pyruvate ImM, final concentrations). Cells were never used at a passage greater than 6. They were seeded into 96- well black walled plates with clear bottom (Fisher Scientific, Loughborough, UK) at a density of 3,000 cells per well in a volume of 200 μΐ per well. The cells were incubated overnight at 37 °C before full growth media was aspirated. Cells were then washed twice with starve media (DMEM with Na Pyruvate ImM final concentration) and left with the last change of media for 24 hours at 37°C. To profile compounds cells were pre-incubated with the inhibitors diluted in starve media with 0.1% fatty acid free BSA (Sigma) and DMSO (0.4% final concentration) for lh prior to the addition of the LPA. LPA diluted in starve media (DMEM with Na Pyruvate ImM final concentration) with 0.1% fatty acid free BSA (Sigma) was added to the cells at a concentration equivalent to the EC50 value. The cells were stimulated with LPA for a period of 24h.
[0334] The proliferation assay was carried out using the DELFIA cell proliferation kit
(Perkin Elmer, Beaconsfield, UK) following the manufacturer's instructions. In brief: BrdU labeling medium was diluted 1 : 100 in starve medium to 100 μΜ, and 20 μΐ was then added per well (except to at least 3 cell containing wells to which 20 μΐ medium was added so as to monitor background/non-specific binding of the anti-BrdU antibody). BrdU was added 20h after the addition of LPA for a period of 4h. The media was then aspirated before the addition of 100 μΐ per well of cell fix solution. After 30 min at room temperature fix solution was removed and ΙΟΟμΙ per well anti-BrdU-Eu working solution (0.5 μg/ml in assay buffer) was added to each well. After 90 min at room temperature the plate was washed 4X with 200 μΐ/well of wash solution and 200 μΐ/well of DELFIA inducer was added. The plate was placed on a shaker at 300 rpm for 15 min at RT before time -resolved fluorescence was measured on the En Vision 2103 Multilabel plate reader (Perkin Elmer), the excitation wave length was 340 nm (UV) and the emission was 615 nm (Europium).
Biological Data
[0335] Table 2 summarizes IC50 values for the compounds of the invention in LPA1 FLIPR, GTPyS and HLF assays. The compounds of the invention have an IC50 within the range of <0.01 μΜ to 2 μΜ in any of the assays described above (reported as an average of all listed data on a single selected batch); LPA1 FLIPR and GTPyS IC50 data are from Method 1 unless otherwise indicated. In particular embodiments, the compounds of the invention have an IC50 in the GTPyS assay within the range of <0.01 μΜ to 1 μΜ; and in more particular embodiments, the compounds of the invention also have an IC50 in the HLF assay within the range of <0.01 μΜ to 1 μΜ, especially from 0.01 μΜ to 0.1 μΜ.
Table 2




[0336] Tables 3 A and 3B compare the activity of a compound of the invention (Example la
and Example 6 and 1 1 , respectively) with various compounds, wherein the

core has been replaced with a different heteroaryl or attached to adjacent atoms at different positions. LPAl FLIPR and GTPyS IC50 data are from Method 1 unless otherwise indicated.



[0338] Table 4 shows the blood concentrations of compound 6a and compound 11 in hepatic portal and jugular vein after oral dosing in cassette format. As shown in Table 4, compound 6a exhibited a higher absorption across the gastrointestinal tract (HPV Cmax = 1015 nM) when compared to compound 11 (HPV Cmax = 327 nM), and showed reasonable metabolic stability (HPV AUC 3804nM.h v Systemic AUC 3715nM.h).
Table 4

[0339] Table 5 summarizes IC50 values for other racemates and stereoisomers prepared following analogous procedures as described in Example 1.
Table 5

Cpd # Structure ICso (μΜ) ICso (μΜ ICso (μΜ)
(method 2)
46
5.93 4.32
47
30
48
30
49
30 30
50
16 6.6
Claims
1. A comp


Z1 and Z2 are independently CH or N; or Z2 is C if attached to R4a;
R1 is hydrogen, halo, hydroxy or C02H;
R2 is hydrogen, halo or methyl; or
R1 and R2 together form cyclopropyl, or a monocyclic 4-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O and S; wherein said 4-6 membered ring is unsubstituted or substituted by 1-2 R9 groups;
R3 is -CO2H, -C(0)NR-(CRR')o-2-CN, -C(0)NRS02R10 or -NR-C(0)R10;
R4a and R4b are independently hydrogen, halo, Ci_6 alkyl, Ci_6 alkoxy or Ci_6 hydroxyalkyl;
R5 is methyl or ethyl;
R6 is hydrogen, Ci_6 alkyl, -(CRR')i_2-0(Ci_4 alkyl), C2^ alkynyl or cyclopropyl;
R6a is hydrogen or Ci_6 alkyl;
R6b is Ci_6 alkyl; cyclopropyl; phenyl substituted with R7 and R8; or a 5-6 membered heterocycle comprising 1-2 heteroatoms selected from N, O or S;
R7 and R8 are independently hydrogen, Ci^ alkyl or halo;
R9 is -C(0)Rn, -C(0)ORn or S02Rn;
R10 and R11 are independently Ci^ alkyl;
R and R' are independently hydrogen or Ci_6 alkyl;
m and n are independently 1-4; and
p is 0-2; or a stereoisomer or pharmaceutically acceptable salt thereof.
2. The compound of claim 1, wherein R6b is isopropyl; methyl; cyclopropyl; phenyl substituted with R7 and R8; tetrahydrofuranyl or tetrahydropyranyl; wherein R7 and R8 are as defined in claim 1.
3. The compound of claim 1, wherein said compound is of Formula (2):

wherein R1, R2, R3, R4a, R4b, R5, R6, R7, R8, Z2, m, n and p are as defined in claim 1.
4. The compound of any one of claims 1-3, wherein R1 and R2 are independently hydrogen or halo; or R1 and R2 together form cyclopropyl, oxetanyl, tetrahydropyanyl or piperidinyl substituted with 1-2 R9 groups, wherein R9 is as defined in claim 1.
5. The compound of any one of claims 1-4, wherein R5 is methyl; and R6 is hydrogen or methyl.
6. The compound of any one of claims 1-5, wherein said compound is of Formula
(3):  wherein R1 and R2 are independently hydrogen or fluoro; or
wherein R1 and R2 are independently hydrogen or fluoro; or
R1 and R2 together form cyclopropyl;
R3, R4a, R4b, R7, R8, m, n and p are as defined in claim 1 ; or
a stereoisomer or pharmaceutically acceptable salt thereof.
7. The compound of claim 6, wherein said compound is of Formula (4):

or a stereoisomer or pharmaceutically acceptable salt thereof.
8. The compound of any one of claims 1-7, wherein R4a is hydrogen, halo, Ci_6 alkyl, Ci_6 alkoxy or Ci_6 hydroxy alkyl; and R4b is hydrogen, halo or Ci_6 alkyl.
9. The compound of any one of claims 1-7, wherein m and n are 1; and R4a and R' are hydrogen.
10. The compound of any one of claims 1-9, wherein R3 is-C02H.
11. The compound of any one of claims 1-10, wherein R7 and R8 are independently hydrogen or halo.
12 The compound of any one of claims 1-11, wherein said compound is selected from:
2· (4- { 4- [5 -( { [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- y lij]phenyl }phenyl)acetic acid;
2- (4- { 4- [5 -( { [( 1 R)- 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1-methyl- 1 H-pyrazol-4- y lij]phenyl }phenyl)acetic acid;
2- (4- { 4- [5 -( { [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- y lij]phenyl }phenyl)acetic acid;
2- (4- { 4- [5 -( { [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -ethyl- 1 H-pyrazol-4- y lij]phenyl }phenyl)acetic acid;
4- {4-[5-({[(lR)-l-(2-chlorophenyl)ethoxy]carbonyl} amino)- 1-methyl- lH-pyrazol-4- y lij]phenyl } benzoic acid;
4- { 4- [5 -( { [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1-methyl- lH-pyrazol-4- y lij]phenyl } benzoic acid;
2- (4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl} amino)- lH-pyrazol-4- y lij]phenyl }phenyl)acetic acid;
2- (4- { 4- [ l-methyl-5-( { [ l-phenylethoxy]carbonyl } amino)- lH-pyrazol-4- y lij]phenyl }phenyl)acetic acid;
2- { 4- [4-( 1 -methyl-5- { [( 1 -phenylpropoxy)carbonyl] amino } - lH-pyrazol-4- y li))phenyl Jphenyl} acetic acid;
2· (4- {4-[ 1 -methyl-5-( { [ l-(2-methylphenyl)ethoxy]carbonyl } amino)- lH-pyrazol-4- y lij]phenyl }phenyl)acetic acid;
2- { 4- [4-(5 - { [( 1 -cyclopropylethoxy)carbonyl] amino } - 1 -methyl- lH-pyrazol-4- y li))phenyl Jphenyl} acetic acid;
2- (4- { 4- [5 -( { [( 1 R)- 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1-methyl- 1 H-pyrazol-4- y lij]phenyl } cyclohexyl) acetic acid ;
2- (4- { 4- [5 -( { [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yljphenyl } cyclohexyl) acetic acid;
2-(4-{4-[5-({ [(lR)-l-(2-chloro-4-fluorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH- pyrazol-4-yl]phenyl } cyclohexyl) acetic acid;
2- (4-{4-[5-({ [l-(2-chloro-4-fluorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH- pyrazol-4-yl]phenyl } cyclohexyl) acetic acid;
3- (4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl} amino)- lH-pyrazol-4- yl]phenyl}phenyl)oxetane-3-carboxylic acid;
3- (4- { 4-[ 1 -methyl-5-( { [ l-phenylethoxy]carbonyl } amino)- lH-pyrazol-4- yl]phenyl}phenyl)oxetane-3-carboxylic acid;
1- [(tert-butoxy)carbonyl]-4-(4- { 4-[ 1 -methyl-5-( { [( 1R)- 1 - phenylethoxy]carbonyl}amino)-lH-pyrazol-4-yl]phenyl}phenyl)piperidine-4-carboxylic acid;
1- [(tert-butoxy)carbonyl]-4-(4- { 4-[ 1 -methyl-5-( { [ 1 -phenylethoxyjcarbonyl } amino)- 1H- pyrazol-4-yl]phenyl}phenyl)piperidine-4-carboxylic acid;
4- (4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl} amino)- lH-pyrazol-4- yljphenyl }phenyl)oxane-4-carboxylic acid;
4-(4- { 4- [ l-methyl-5-( { [ l-phenylethoxy]carbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)oxane-4-carboxylic acid;
( 1 R)- 1 -phenylethyl N- [4-(4- { 4- [(methanesulfonylcarbamoyl)methyl]phenyl }phenyl)- 1 - methyl- 1 H-pyrazol- 5 -yl] carbamate ;
1-phenylethyl N- [4-(4- { 4-[(methanesulfonylcarbamoyl)methyl]phenyl }phenyl)- 1 - methyl- 1 H-pyrazol- 5 -yl] carbamate ;
(R)-l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid;
l-(4'-(l-methyl-5-((l-phenylethoxy)carbonylamino)-lH-pyrazol-4-yl)biphenyl-4- yl)cyclopropanecarboxylic acid;
l-(4- { 4- [5-( { [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- lH-pyrazol-4- yljphenyl }phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{2-fluoro-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl }phenyl)cyclopropane- 1 -carboxylic acid;
1 - (4- { 2-fluoro-4- [ 1 -methyl- 5 - ( { [ 1 -pheny lethoxy ] carbonyl } amino) - 1 H-pyrazol-4- yljphenyl }phenyl)cyclopropane- 1 -carboxylic acid; 2-(4- { 4- [5 -( { [( 1 R)- 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yl]phenyl}phenyl)-2,2-difluoroacetic acid;
2- (4-{4-[5-({ [l-(2-chlorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH-pyrazol-4- yl]phenyl}phenyl)-2,2-difluoroacetic acid;
l-(4-{3-methyl-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 -(4- { 3 -methyl-4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
3- (4-{4-[5-({ [(l R)- 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yl]phenyl}phenyl)propanoic acid;
3 -(4- { 4- [5 -( { [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yl]phenyl}phenyl)propanoic acid;
1- (2-fluoro-4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 -(2-fluoro-4- { 4- [ 1 -methyl-5-( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
2- (2-fluoro-4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2-(2-fluoro-4-{4-[l-methyl-5-({ [l-phenylethoxy]carbonyl} amino)- lH-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2-(2-chloro-4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2-(2-chloro-4- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2-(2-methyl-4- { 4- [ 1 -methyl-5 -( { [( 1R)- 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2-(2-methyl-4- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)acetic acid;
2,2-difluoro-2-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl} amino)- lH-pyrazol- 4-yl]phenyl}phenyl)acetic acid;
2,2-difluoro-2-(4- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)acetic acid; l-(4-{3-fluoro-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 -(4- { 3 -fluoro-4- [ 1 -methyl-5-( { [ 1 -phenylethoxy] carbonyl } amino)- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 -(4- { 2-methyl-4- [ 1 -methyl-5 -( { [( 1R)- 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 -(4- { 2-methyl-4- [ 1 -methyl-5 -( { [ 1 -phenylethoxy] carbonyl } amino)- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
3-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)propanoic acid;
3-(4-{4-[l-methyl-5-({ [l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)propanoic acid;
l-(4-{3-methoxy-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol- 4-yl]phenyl}phenyl)cyclopropane- 1-carboxylic acid;
1 -(4- { 3-methoxy-4-[ 1 -methyl-5-({ [ 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{2-methoxy-4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol- 4-yl]phenyl}phenyl)cyclopropane- 1-carboxylic acid;
1 -(4- {2-methoxy-4-[ 1 -methyl-5-({ [ 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 -methyl- 5 - { 4- [ 1 -methyl- 5 - ( { [( 1 R)- 1 -phenylethoxy ] c arbonyl } amino) - 1 H-pyrazol-4- yljphenyl} -2,3-dihydro- IH-indene- 1-carboxylic acid;
1 -methyl- 5 - { 4- [ 1 -methyl- 5 - ( { [ 1 -phenylethoxy ] carbonyl } amino) - 1 H-pyrazol-4- yljphenyl} -2,3-dihydro- IH-indene- 1-carboxylic acid;
6- { 4- [5-({ [ 1 -(2-chlorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- lH-pyrazol-4- yljphenyl} -2,3-dihydro- IH-indene- 1-carboxylic acid;
l-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}arnino)-lH-l,2,3-triazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{4-[l-methyl-5-({ [l-phenylethoxy]carbonyl}amino)-lH-l,2,3-triazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{4-[l-methyl-5-({ [(lR)-l-phenylpropoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid; l-(4-{4-[l-methyl-5-({ [l-phenylpropoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 - { 4- [4- ( 1 -methyl- 5 - { [( 1 -pheny lbutoxy )carbonyl] amino } - 1 H-pyrazol-4- yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
1 - { 4- [4-(5 - { [(2,2-dimethyl- 1 -pheny lpropoxy)carbonyl] amino } - 1 -methyl- lH-pyrazol-4- yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
1 -(4- {4-[ 1 -methyl-5-( { [ l-(3-methylphenyl)ethoxy]carbonyl } amino)- lH-pyrazol-4- yljphenyl } pheny l)cyclopropane- 1 -carboxylic acid;
1- (4-{4-[5-({ [(lR)-l-(2-chloro-4-fluorophenyl)ethoxy]carbonyl}amino)-l-methyl-lH- pyrazol-4-yl]phenyl}phenyl)cyclopropane-l -carboxylic acid;
1 -(4- { 4- [5 -( { [ 1 -(2-chloro-4-fluorophenyl)ethoxy]carbonyl } amino)- 1 -methyl- 1H- pyrazol-4-yl]phenyl}phenyl)cyclopropane-l -carboxylic acid;
1 - { 4- [4-(5 - { [(tert-butoxy)carbonyl] amino } - 1 -methyl- 1 H-pyrazol-4- yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
2- acetamido-3-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl} amino)- lH-pyrazol- 4-yl]phenyl} pheny l)propanoic acid;
2-acetamido-3-(4-{4-[l-methyl-5-({ [l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl} pheny l)propanoic acid;
2-hydroxy-2-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl} pheny l)propanoic acid;
2-hydroxy-2-(4-{4-[l-methyl-5-({ [1-phenylethoxy] carbonyl} amino)- lH-pyrazol-4- yljphenyl} pheny l)propanoic acid;
l-(4-{6-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4-yl]pyridin- 3-yl}phenyl)cyclopropane-l-carboxylic acid;
1 -(4- { 6-[ 1 -methyl-5-( { [ l-phenylethoxy]carbonyl } amino)- lH-pyrazol-4-yl]pyridin-3- yl } pheny l)cyclopropane- 1 -carboxylic acid;
1 - { 4- [2-(hydroxymethyl)-4- [ l-methyl-5-( { [( 1R)- 1 -phenylethoxyjcarbonyl } amino)- 1H- pyrazol-4-yl]phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
1 - { 4- [2-(hydroxymethyl)-4- [ l-methyl-5-( { [ 1 -phenylethoxyjcarbonyl } amino)- 1H- pyrazol-4-yl]phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
l-(6-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}pyridin-3-yl)cyclopropane-l -carboxylic acid; 1 -(6- { 4- [ 1 -methyl-5-( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}pyridin-3-yl)cyclopropane-l-carboxylic acid;
6- { 4- [ 1 -methy 1-5 -( { [ ( 1 R) - 1 -phenylethoxy ] c arbonyl } amino) - 1 H-pyrazol-4- yljphenyl } spiro [2.5 Joctane- 1 -carboxylic acid;
6- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- lH-pyrazol-4- yljphenyl } spiro [2.5 Joctane- 1 -carboxylic acid;
1 - { 4- [4-( 1 -methyl-5- { [( 1 -phenyl- 1 -deuteroethoxy)carbonyl] amino } - 1 H-pyrazol-4- yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
1 - { 4- [4- ( 1 -methyl- 5 - { [( 1 -phenyl- 1 -deutero- 1 - trideuteromethylmethoxy)carbonyl] amino } - 1 H-pyrazol-4-yl)phenyl]phenyl } cyclopropane- 1 - carboxylic acid;
1 - { 4- [4- ( 1 -methyl- 5 - { [( 1 -pentadeuterophenyl- 1 -deutero- 1 - trideuteromethylmethoxy)carbonyl] amino } - 1 H-pyrazol-4-yl)phenyl]phenyl } cyclopropane- 1 - carboxylic acid;
1 - { 4- [4-(5 - { [(2-methoxy- 1 -phenylethoxy)carbonyl]amino } - 1 -methyl- lH-pyrazol-4- yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
l-(4-{4-[l-methyl-5-({ [(l-phenylbut-2-yn-l-yl)oxy]carbonyl} amino)- lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 - { 4- [4-(5 - { [(3-methoxy- 1 -phenylpropoxy)carbonyl] amino } - 1 -methyl- 1 H-pyrazol-4- yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
1 -(4- { 4- [5 -( { [cyclopropyl(phenyl)methoxy]carbonyl } amino)- 1 -methyl- 1 H-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 - { 4- [4-(5 - { [(2-ethoxy- 1 -phenylethoxy)carbonyl] amino } - 1 -methyl- 1 H-pyrazol-4- yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
l-[4-(4-{ l-methyl-5-[({[(2R)-3-methylbutan-2-yl]oxy}carbonyl)amino]-lH-pyrazol-4- yl } phenyl)phenyl]cyclopropane- 1 -carboxylic acid;
l-[4-(4-{ l-methyl-5-[({[(2S)-3-methylbutan-2-yl]oxy}carbonyl)amino]-lH-pyrazol-4- yl } phenyl)phenyl]cyclopropane- 1 -carboxylic acid;
1 -[4-(4- { 1 -methyl-5- [( { [3-methylbutan-2-yl]oxy } carbonyl)amino]- lH-pyrazol-4- yl } phenyl)phenyl]cyclopropane- 1 -carboxylic acid;
1 - { 4- [4-(5 - { [( 1 -cyclopropylethoxy)carbonyl] amino } - 1 -methyl- lH-pyrazol-4- yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid; l-(4-{4-[l-methyl-5-({ [l-(2-methylphenyl)ethoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{4-[l-methyl-5-({ [l-(oxan-4-yl)ethoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-(4-{4-[l-methyl-5-({ [l-(oxolan-3-yl)ethoxy]carbonyl}amino)-lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
1 - { 4- [4-(5 - { [(benzyloxy)carbonyl] amino } - 1 -methyl- 1 H-pyrazol-4- yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
l-(4- { 4-[ 1 -methyl-5-( { [(2-methylphenyl)methoxy]carbonyl } amino)- lH-pyrazol-4- yljphenyl } phenyl)cyclopropane- 1 -carboxylic acid;
l-{4-[4-(l-methyl-5-{ [(oxan-4-ylmethoxy)carbonyl]amino}-lH-pyrazol-4- yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
l-{4-[4-(l-methyl-5-{ [(oxolan-3-ylmethoxy)carbonyl]amino}-lH-pyrazol-4- yl)phenyl]phenyl } cyclopropane- 1 -carboxylic acid;
l-[6-(4-{ l-methyl-5-[({[(2R)-3-methylbutan-2-yl]oxy}carbonyl)amino]-lH-pyrazol-4- yl}phenyl)pyridin-3-yl]cyclopropane-l-carboxylic acid;
l-[6-(4-{ l-methyl-5-[({[3-methylbutan-2-yl]oxy}carbonyl)amino]-lH-pyrazol-4- yl}phenyl)pyridin-3-yl]cyclopropane-l-carboxylic acid;
l-{6-[4-(5-{[( 1 -cyclopropylethoxy)carbonyl] amino } - 1 -methyl- lH-pyrazol-4- yl)phenyl]pyridin-3 -yl } cyclopropane- 1 -carboxylic acid;
l-(6-{4-[l-methyl-5-({ [l-(oxolan-3-yl)ethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}pyridin-3-yl)cyclopropane-l -carboxylic acid;
1 - { 6- [3 -methoxy-4-( 1 -methyl-5- { [( 1 -phenyl- 1 -deutero- 1 - trideuteromethylmethoxy)carbonyl]amino}-lH-pyrazol-4-yl)phenyl]pyridin-3-yl}cyclopropane- 1 -carboxylic acid;
l-(6-(3-methoxy-4-(l-methyl-5-(((l-phenylethoxy)carbonyl)amino)-lH-pyrazol-4- yl)phenyl)pyridin-3-yl)cyclopropanecarboxylic acid;
l-acetyl-4-(4-{4-[l-methyl-5-({ [(lR)-l-phenylethoxy]carbonyl}amino)-lH-pyrazol-4- yl]phenyl}phenyl)piperidine-4-carboxylic acid;
1 -acetyl-4-(4- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxyjcarbonyl } amino)- 1 H-pyrazol-4- yl]phenyl}phenyl)piperidine-4-carboxylic acid; 1 -methanesulfonyl-4-(4- { 4- [ 1 -methyl-5 -( { [( 1R)- 1 -phenylethoxyjcarbonyl } amino)- 1 H- pyrazol-4-yl]phenyl}phenyl)piperidine-4-carboxylic acid;
1 -methanesulfonyl-4-(4- { 4- [ 1 -methyl-5 -( { [ 1 -phenylethoxy] carbonyl } amino)- 1H- pyrazol-4-yl]phenyl}phenyl)piperidine-4-carboxylic acid;
( 1 R)- 1 -phenylethyl N- { 4- [4-(4- { 1 - [(2-cyanoethyl)carbamoyl]cyclopropyl }
phenyl)phenyl] - 1 -methyl- 1 H-pyrazol-5 -yl } carbamate; and
1 -phenylethyl N- { 4- [4-(4- { 1 - [(2-cyanoethyl)carbamoyl]cyclopropyl } phenyl)phenyl] - 1 - methyl- 1 H-pyrazol-5 -yl } carbamate; or
a stereoisomer or pharmaceutically acceptable salt thereof.
13. A pharmaceutical composition comprising a therapeutically effective amount of a compound of any one of claims 1-12 or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
14. A combination comprising a compound of any one of claims 1-12 or a pharmaceutically acceptable salt thereof, and a second therapeutic agent selected from corticosteroids, immunosuppresant, analgesics, anti-cancer agent, anti-inflammatories, chemokine receptor antagonists, bronchodilators, leukotriene receptor antagonists, leukotriene formation inhibitors, monoacylglycerol kinase inhibitors, phospholipase A] inhibitors, phospholipase A2 inhibitors, and lysophospholipase D (lysoPLD) inhibitors, autotaxin inhibitors, decongestants, antihistamines, mucolytics, anticholinergics, antitussives, expectorants, and β-2 agonists.
15. A method for treating a LPA-dependent or LPA-mediated disease or condition comprising administering to a subject in need thereof a therapeutically effective amount of a compound of any one of claims 1-12 or a pharmaceutically acceptable salt thereof, and optionally in combination with a second therapeutic agent.
16. A compound according to any one of claims 1-12 or a pharmaceutically acceptable salt thereof, for use in the treatment of a LPA-dependent or LPA-mediated disease or condition.
Use of a compound of any one of claims 1-12 or a pharmaceutically acceptable salt thereof, for the preparation of a medicament for the treatment of a LPA-dependent or LPA- mediated disease or condition in a subject.
18. The method of claim 15, the compound of claim 16, or the use of claim 17, wherein said LPA-dependent or LPA-mediated disease or condition is selected from: (i) pulmonary fibrosis, idiopathic pulmonary fibrosis, iatrogenic drug-induced fibrosis, occupational or environmental induced fibrosis, sarcoidosis, hypersensitivity pneumonia, collagen vascular disease, alveolar proteinosis, langerhans cell granulomatosis,
lymphangioleiomyomatosis, Hermansky-Pudlak Syndrome, tuberous sclerosis,
neurofibromatosis, metabolic storage disorders and familial interstitial lung disease; (ii) fibrosis of organs or tissues, scarring, liver diseases, dermatological conditions, cancer, cardiovascular diseases, respiratory diseases or conditions, inflammatory diseases, gastrointestinal tract diseases, renal diseases, urinary tract-associated diseases, inflammatory diseases of lower urinary tract, dysuria, frequent urination, pancreas disease, arterial obstruction, cerebral infarction, cerebral hemorrhage, pain, peripheral neuropathy, and fibromyalgia; (iii) radiation induced fibrosis; chronic obstructive pulmonary disease (COPD), scleroderma, systemic sclerosis, bleomycin induced pulmonary fibrosis, chronic asthma, silicosis, asbestos induced pulmonary fibrosis, acute respiratory distress syndrome (ARDS), kidney fibrosis,
tubulointerstitium fibrosis, glomerular nephritis, focal segmental glomerular sclerosis, lupus nephritis, IgA nephropathy, hypertension, Alport, gut fibrosis, liver fibrosis, cirrhosis, alcohol induced liver fibrosis, toxic/drug induced liver fibrosis, hemochromatosis, nonalcoholic steatohepatitis (NASH), biliary duct injury; primary biliary cirrhosis, infection induced liver fibrosis, viral induced liver fibrosis, autoimmune hepatitis, corneal scarring, hypertrophic scarring, Dupuytren's disease, keloids, cutaneous fibrosis, cutaneous scleroderma, spinal cord injury/fibrosis, myelofibrosis, vascular restenosis, atherosclerosis, arteriosclerosis, Wegener's granulomatosis, Peyronie's disease, chronic lymphocytic leukemia, tumor metastasis, transplant organ rejection, endometreosis, neonatal respiratory distress syndrome and neuropathic pain; or (iv) renal fibrosis, acute kidney injury, chronic kidney disease, skin fibrosis, fibrosis of the gut, ocular fibrosis, breast cancer, pancreatic cancer, ovarian cancer, prostate cancer, glioblastoma, bone cancer, colon cancer, bowel cancer, head and neck cancer, melanoma, multiple myeloma, chronic lymphocytic leukemia, cancer pain, tumor metastatis, transplant organ rejection, age related macular degeneration (AMD), diabetic retinopathy, and Raynaud's phenomenon.
19. The method of claim 15, the compound of claim 16, or the use of claim 17, wherein said LPA-dependent or LPA-mediated disease or condition is selected from pulmonary fibrosis, idiopathic pulmonary fibrosis, asthma, chronic obstructive pulmonary disease (COPD), renal fibrosis, acute kidney injury, chronic kidney disease, liver fibrosis, skin fibrosis, fibrosis of the gut, ocular fibrosis, breast cancer, pancreatic cancer, ovarian cancer, prostate cancer, glioblastoma, bone cancer, colon cancer, bowel cancer, head and neck cancer, melanoma, multiple myeloma, chronic lymphocytic leukemia, cancer pain, tumor metastatis, transplant organ rejection, scleroderma, age related macular degeneration (AMD), diabetic retinopathy, collagen vascular disease, atherosclerosis, Raynaud's phenomenon and neuropathic pain.
20. The method of claim 15, the compound of claim 16, or the use of claim 17, wherein said LPA-dependent or LPA-mediated disease or condition is idiopathic pulmonary fibrosis.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161472547P | 2011-04-06 | 2011-04-06 | |
| US61/472,547 | 2011-04-06 | ||
| US201261607393P | 2012-03-06 | 2012-03-06 | |
| US61/607,393 | 2012-03-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012138648A1 true WO2012138648A1 (en) | 2012-10-11 |
Family
ID=45953302
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/031985 WO2012138648A1 (en) | 2011-04-06 | 2012-04-03 | Compositions and methods for modulating lpa receptors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2012138648A1 (en) |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013025733A1 (en) * | 2011-08-15 | 2013-02-21 | Intermune, Inc. | Lysophosphatidic acid receptor antagonists |
| WO2013189864A1 (en) * | 2012-06-20 | 2013-12-27 | F. Hoffmann-La Roche Ag | N-alkyltriazole compounds as lpar antagonists |
| WO2013189862A1 (en) * | 2012-06-20 | 2013-12-27 | F. Hoffmann-La Roche Ag | Substituted pyrazole compounds as lpar antagonists |
| WO2013189865A1 (en) * | 2012-06-20 | 2013-12-27 | F. Hoffmann-La Roche Ag | N-aryltriazole compounds as lpar antagonists |
| WO2014104372A1 (en) | 2012-12-28 | 2014-07-03 | 宇部興産株式会社 | Halogen-substituted heterocyclic compound |
| WO2014113485A1 (en) * | 2013-01-15 | 2014-07-24 | Intermune, Inc. | Lysophosphatidic acid receptor antagonists |
| WO2014145873A3 (en) * | 2013-03-15 | 2014-12-18 | Epigen Biosciences, Inc. | Heterocyclic compounds useful in the treatment of disease |
| WO2015199234A1 (en) * | 2014-06-27 | 2015-12-30 | 宇部興産株式会社 | Salt of halogen-substituted heterocyclic compound |
| WO2016004166A1 (en) * | 2014-07-02 | 2016-01-07 | Xavier University Of Louisiana | Boron-based prodrug strategy for increased bioavailability and lower-dosage requirements for drug molecules containing at least one phenol (or aromatic hydroxyl) group |
| WO2017086430A1 (en) * | 2015-11-20 | 2017-05-26 | 宇部興産株式会社 | Pharmaceutical composition for treatment or prophylaxis of nash |
| WO2019046239A1 (en) * | 2017-09-04 | 2019-03-07 | Eli Lilly And Company | Lysophosphatidic acid receptor 1 (lpar1) inhibitor compounds |
| WO2019126090A1 (en) * | 2017-12-19 | 2019-06-27 | Bristol-Myers Squibb Company | Triazole n-linked carbamoyl cyclohexyl acids as lpa antagonists |
| WO2019126094A1 (en) * | 2017-12-19 | 2019-06-27 | Bristol-Myers Squibb Company | Cyclohexyl acid triazole azoles as lpa antagonists |
| WO2019126093A1 (en) * | 2017-12-19 | 2019-06-27 | Bristol-Myers Squibb Company | Cyclohexyl acid triazole azines as lpa antagonists |
| WO2019246109A1 (en) | 2018-06-18 | 2019-12-26 | Epigen Biosciences, Inc. | Heterocyclic compounds useful in the treatment of disease |
| CN111434653A (en) * | 2019-01-15 | 2020-07-21 | 武汉朗来科技发展有限公司 | Triazole compound and preparation method and application thereof |
| WO2020257135A1 (en) * | 2019-06-18 | 2020-12-24 | Bristol-Myers Squibb Company | Triazole carboxylic acids as lpa antagonists |
| JP2021507898A (en) * | 2017-12-19 | 2021-02-25 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Pyrazole cyclohexylate as an LPA antagonist |
| WO2021097039A1 (en) * | 2019-11-15 | 2021-05-20 | Gilead Sciences, Inc. | Triazole carbamate pyridyl sulfonamides as lpa receptor antagonists and uses thereof |
| EA038294B1 (en) * | 2013-05-24 | 2021-08-05 | Эпиджен Байосайенсиз, Инк. | Heterocyclic compounds useful in the treatment of diseases |
| US11180488B2 (en) | 2017-12-19 | 2021-11-23 | Bristol-Myers Squibb Company | Isoxazole o-linked carbamoyl cyclohexyl acids as LPA antagonists |
| WO2022034568A1 (en) * | 2020-08-11 | 2022-02-17 | Viva Star Biosciences Limited | Triazole-pyridinyl substituted azacyclohexyl acetic acid compounds as lpa receptor antagonists |
| US11261174B2 (en) | 2017-12-19 | 2022-03-01 | Bristol-Myers Squibb Company | Pyrazole O-linked carbamoyl cyclohexyl acids as LPA antagonists |
| US11261180B2 (en) | 2017-12-19 | 2022-03-01 | Bristol-Myers Squibb Company | Cyclohexyl acid isoxazole azoles as LPA antagonists |
| US11312706B2 (en) | 2017-12-19 | 2022-04-26 | Bristol-Myers Squibb Company | Cyclohexyl acid pyrazole azines as LPA antagonists |
| US11319309B2 (en) | 2017-12-19 | 2022-05-03 | Bristol-Myers Squibb Company | Cyclohexyl acid isoxazole azines as LPA antagonists |
| US11384067B2 (en) | 2017-12-19 | 2022-07-12 | Bristol-Myers Squibb Company | Pyrazole n-linked carbamoyl cyclohexyl acids as LPA antagonists |
| US11447475B2 (en) | 2017-12-19 | 2022-09-20 | Bristol-Myers Squibb Company | Isoxazole N-linked carbamoyl cyclohexyl acids as LPA antagonists |
| WO2022240879A1 (en) * | 2021-05-11 | 2022-11-17 | Gilead Sciences, Inc. | Lpa receptor antagonists and uses thereof |
| US11584738B2 (en) | 2020-06-03 | 2023-02-21 | Gilead Sciences, Inc. | LPA receptor antagonists and uses thereof |
| US11702407B2 (en) | 2020-06-03 | 2023-07-18 | Gilead Sciences, Inc. | LPA receptor antagonists and uses thereof |
| US11884627B2 (en) | 2022-02-25 | 2024-01-30 | Lhotse Bio, Inc. | Compounds and compositions for treating conditions associated with LPA receptor activity |
| WO2024022314A1 (en) * | 2022-07-25 | 2024-02-01 | 武汉人福创新药物研发中心有限公司 | Triazole compounds and use thereof as lpar1 antagonist |
| US11939318B2 (en) | 2021-12-08 | 2024-03-26 | Gilead Sciences, Inc. | LPA receptor antagonists and uses thereof |
| CN114728168B (en) * | 2019-11-15 | 2024-04-09 | 吉利德科学公司 | Triazole carbamate pyridylsulfonamides as LPA receptor antagonists and their use |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0039051A2 (en) | 1980-04-24 | 1981-11-04 | Merck & Co. Inc. | Mannich-base hydroxamic acid prodrugs for the improved bioavailability of non-steroidal anti-inflammatory agents, a process for preparing and a pharmaceutical composition containing them |
| US5824683A (en) | 1995-11-28 | 1998-10-20 | Schering Corporation | 2'- 4'-halo- 1,1'-biphenyl!-4-yl!methyl!-5'-methyl-spiro cyclopentane-1,7' (8'H)- 3H! imidazo 2,1-b!purin!-4' (5'H)-ones |
| WO2004078163A2 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2011017350A2 (en) * | 2009-08-04 | 2011-02-10 | Amira Pharmaceuticals, Inc. | Compounds as lysophosphatidic acid receptor antagonists |
-
2012
- 2012-04-03 WO PCT/US2012/031985 patent/WO2012138648A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0039051A2 (en) | 1980-04-24 | 1981-11-04 | Merck & Co. Inc. | Mannich-base hydroxamic acid prodrugs for the improved bioavailability of non-steroidal anti-inflammatory agents, a process for preparing and a pharmaceutical composition containing them |
| US5824683A (en) | 1995-11-28 | 1998-10-20 | Schering Corporation | 2'- 4'-halo- 1,1'-biphenyl!-4-yl!methyl!-5'-methyl-spiro cyclopentane-1,7' (8'H)- 3H! imidazo 2,1-b!purin!-4' (5'H)-ones |
| WO2004078163A2 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2011017350A2 (en) * | 2009-08-04 | 2011-02-10 | Amira Pharmaceuticals, Inc. | Compounds as lysophosphatidic acid receptor antagonists |
Non-Patent Citations (32)
| Title |
|---|
| "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", 2002, WILEY-VCH |
| "Methods of Organic Chemistry", vol. 15/1, 1974, GEORG THIEME VERLAG, article "Methoden der organischen Chemie" |
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY |
| "Remington's Pharmaceutical Sciences", 1990, MACK PRINTING COMPANY, pages: 1289 - 1329 |
| "The Peptides", vol. 3, 1981, ACADEMIC PRESS |
| "The Practice of Medicinal Chemistry", 2001, ACADEMIC PRESS |
| BOUCHARABA ET AL., PROC. NATL. ACAD. SCI. U. S. A., 2006 |
| BOURGOIN; ZHAO, CURR. OPIN. INVESTIG. DRUGS., 2010 |
| BUNDGAARD, J., MED. CHEM., vol. 2503, 1989 |
| BUNDGAARD: "Design of Prodrugs", 1985, ELSEVIER |
| CHOI ET AL., ANNU. REV. PHARMACOL. TOXICOL., 2010 |
| CHOI ET AL., ANNU. REV. PHARMACOL. TOXICOL., vol. 50, 2010, pages 157 - 86 |
| H.-D. JAKUBKE; H. JESCHKEIT: "Amino acids, Peptides, Proteins", 1982, VERLAG CHEMIE, article "Aminosauren, Peptide, Proteine" |
| HOUBEN-WEYL: "Methods of Organic Synthesis", vol. 21, 1952, THIEME |
| INOUE ET AL., CLINICAL SCIENCE, vol. 96, 1999, pages 431 - 436 |
| J. F. W. MCOMIE: "Protective Groups in Organic Chemistry", 1973, PLENUM PRESS |
| J.P. PRADERE ET AL.: "Biochimica et Biophysica Acta", MOLECULAR AND CELL BIOLOGY OF LIPIDS, vol. 1781, no. 9, 2008, pages 582 - 587 |
| JOCHEN LEHMANN: "Chemie der Kohlenhydrate: Monosaccharide und Derivate", 1974, GEORG THIEME VERLAG |
| JS SWANEY ET AL: "A novel, orally active LPA1 receptor antagonist inhibits lung fibrosis in the mouse bleomycin model", BRITISH JOURNAL OF PHARMACOLOGY, vol. 160, no. 7, 1 August 2010 (2010-08-01), pages 1699 - 1713, XP055018263, ISSN: 0007-1188, DOI: 10.1111/j.1476-5381.2010.00828.x * |
| KOH ET AL., J. CLIN. INVEST., vol. 102, 1998, pages 716 - 727 |
| LIU ET AL., CANCER CELL, 2009 |
| NOGUCHI ET AL., CURR. OPIN. PHARMACOL., 2009 |
| P. G. M. WUTS: "Protective Groups in Organic Synthesis", 1999, WILEY |
| PRADERE, J. AM. SOC. NEPHROL., 2007 |
| SWANEY ET AL., BRIT. J. PHARMACOL., vol. 160, 2010, pages 1699 - 1713 |
| TAGER ET AL., NAT. MED., 2008 |
| TAGER ET AL., NATURE MEDICINE, vol. 14, no. 1, 2008, pages 45 - 54 |
| TANAKA ET AL., J. BIOL. CHEM., 2006 |
| TIGYI, BRIT. PHARMACOL. SOC., vol. 161, 2010, pages 241 - 270 |
| TIGYI, BRITISH J PHARMACOLOGY, vol. 161, 2010, pages 241 - 270 |
| TOKUMURA ET AL., J. BIOL. CHEM., 2002 |
| ZHAO ET AL., CELL SIGNAL., 2009 |
Cited By (115)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8975235B2 (en) | 2011-03-20 | 2015-03-10 | Intermune, Inc. | Lysophosphatidic acid receptor antagonists |
| WO2013025733A1 (en) * | 2011-08-15 | 2013-02-21 | Intermune, Inc. | Lysophosphatidic acid receptor antagonists |
| WO2013189864A1 (en) * | 2012-06-20 | 2013-12-27 | F. Hoffmann-La Roche Ag | N-alkyltriazole compounds as lpar antagonists |
| WO2013189862A1 (en) * | 2012-06-20 | 2013-12-27 | F. Hoffmann-La Roche Ag | Substituted pyrazole compounds as lpar antagonists |
| WO2013189865A1 (en) * | 2012-06-20 | 2013-12-27 | F. Hoffmann-La Roche Ag | N-aryltriazole compounds as lpar antagonists |
| US9321738B2 (en) | 2012-06-20 | 2016-04-26 | Hoffman-La Roche Inc. | N-alkyltriazole compounds as LPAR antagonists |
| JP2015520203A (en) * | 2012-06-20 | 2015-07-16 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | N-aryltriazole compounds as LPAR antagonists |
| CN104395299A (en) * | 2012-06-20 | 2015-03-04 | 霍夫曼-拉罗奇有限公司 | N-aryltriazole compounds as lpar antagonists |
| RU2756506C2 (en) * | 2012-12-28 | 2021-10-01 | Убе Индастриз, Лтд. | Halogen-substituted heterocyclic compound |
| WO2014104372A1 (en) | 2012-12-28 | 2014-07-03 | 宇部興産株式会社 | Halogen-substituted heterocyclic compound |
| CN104884447A (en) * | 2012-12-28 | 2015-09-02 | 宇部兴产株式会社 | Halogen-substituted heterocyclic compound |
| KR20150100756A (en) | 2012-12-28 | 2015-09-02 | 우베 고산 가부시키가이샤 | Halogen-substituted heterocyclic compound |
| CN104884447B (en) * | 2012-12-28 | 2017-10-10 | 宇部兴产株式会社 | Halogen substituted heterocyclic compound |
| CN107698555B (en) * | 2012-12-28 | 2020-05-05 | 宇部兴产株式会社 | Halogen-substituted heterocyclic compounds |
| US10597375B2 (en) | 2012-12-28 | 2020-03-24 | Ube Industries, Ltd. | Halogen-substituted heterocyclic compound |
| AU2013366898B2 (en) * | 2012-12-28 | 2017-06-29 | Ube Corporation | Halogen-substituted heterocyclic compound |
| JP2019014739A (en) * | 2012-12-28 | 2019-01-31 | 宇部興産株式会社 | Halogen substituted heterocyclic compound |
| JPWO2014104372A1 (en) * | 2012-12-28 | 2017-01-19 | 宇部興産株式会社 | Halogen-substituted heterocyclic compounds |
| EP3360869A1 (en) | 2012-12-28 | 2018-08-15 | Ube Industries, Ltd. | Halogen-substituted heterocyclic compound useful for the treatment of diseases caused by lpa |
| US10000463B2 (en) | 2012-12-28 | 2018-06-19 | Ube Industries, Ltd. | Halogen-substituted heterocyclic compound |
| RU2649398C2 (en) * | 2012-12-28 | 2018-04-03 | Убе Индастриз, Лтд. | Halogen substituted heterocyclic compound |
| CN107698555A (en) * | 2012-12-28 | 2018-02-16 | 宇部兴产株式会社 | Halogen substituted heterocyclic compound |
| JP2017214410A (en) * | 2012-12-28 | 2017-12-07 | 宇部興産株式会社 | Halogen substituted heterocyclic compound |
| WO2014113485A1 (en) * | 2013-01-15 | 2014-07-24 | Intermune, Inc. | Lysophosphatidic acid receptor antagonists |
| US10526298B2 (en) | 2013-03-15 | 2020-01-07 | Epigen Biosciences, Inc. | Heterocyclic compounds useful in the treatment of disease |
| CN112424174B (en) * | 2013-03-15 | 2024-02-27 | 艾匹根生物技术有限公司 | Heterocyclic compounds useful in the treatment of disease |
| WO2014145873A3 (en) * | 2013-03-15 | 2014-12-18 | Epigen Biosciences, Inc. | Heterocyclic compounds useful in the treatment of disease |
| US10000459B2 (en) | 2013-03-15 | 2018-06-19 | Epigen Biosciences, Inc. | Heterocyclic compounds useful in the treatment of disease |
| KR20150130415A (en) * | 2013-03-15 | 2015-11-23 | 에피젠 바이오싸이언시즈, 아이엔씨. | Heterocyclic compounds useful in the treatment of disease |
| KR102090231B1 (en) * | 2013-03-15 | 2020-03-17 | 에피젠 바이오싸이언시즈, 아이엔씨. | Heterocyclic compounds useful in the treatment of disease |
| US10570103B2 (en) | 2013-03-15 | 2020-02-25 | Epigen Biosciences, Inc. | Heterocyclic compounds useful in the treatment of disease |
| CN112424174A (en) * | 2013-03-15 | 2021-02-26 | 艾匹根生物技术有限公司 | Heterocyclic compounds useful for the treatment of diseases |
| EA033923B1 (en) * | 2013-03-15 | 2019-12-10 | Эпиджен Байосайенсиз, Инк. | Heterocyclic compounds useful in the treatment of diseases |
| AU2014232383B2 (en) * | 2013-03-15 | 2019-01-17 | Epigen Biosciences, Inc. | Heterocyclic compounds useful in the treatment of disease |
| JP2016515536A (en) * | 2013-03-15 | 2016-05-30 | エピゲン バイオサイエンシズ, インコーポレイテッドEpigen Biosciences, Inc. | Heterocyclic compounds useful for the treatment of diseases |
| US11427552B2 (en) | 2013-03-15 | 2022-08-30 | Epigen Biosciences, Inc. | Heterocyclic compounds useful in the treatment of disease |
| EA038294B1 (en) * | 2013-05-24 | 2021-08-05 | Эпиджен Байосайенсиз, Инк. | Heterocyclic compounds useful in the treatment of diseases |
| WO2015199234A1 (en) * | 2014-06-27 | 2015-12-30 | 宇部興産株式会社 | Salt of halogen-substituted heterocyclic compound |
| CN106458964B (en) * | 2014-06-27 | 2019-11-22 | 宇部兴产株式会社 | The salt of halogen substituted heterocyclic compound |
| KR102433588B1 (en) | 2014-06-27 | 2022-08-19 | 우베 가부시키가이샤 | Salt of halogen-substituted heterocyclic compound |
| KR20170016988A (en) | 2014-06-27 | 2017-02-14 | 우베 고산 가부시키가이샤 | Salt of halogen-substituted heterocyclic compound |
| US10023554B2 (en) | 2014-06-27 | 2018-07-17 | Ube Industries, Ltd. | Halogen-substituted heterocyclic compound salt |
| JPWO2015199234A1 (en) * | 2014-06-27 | 2017-04-27 | 宇部興産株式会社 | Salts of halogen-substituted heterocyclic compounds |
| CN106458964A (en) * | 2014-06-27 | 2017-02-22 | 宇部兴产株式会社 | Salt of halogen-substituted heterocyclic compound |
| CN106715446A (en) * | 2014-07-02 | 2017-05-24 | 路易斯安那泽维尔大学 | Boron-based prodrug strategy for increased bioavailability and lower-dosage requirements for drug molecules containing at least one phenol (or aromatic hydroxyl) group |
| US10112962B2 (en) | 2014-07-02 | 2018-10-30 | Xavier University | Boron-based prodrug strategy for increased bioavailability and lower-dosage requirements for drug molecules containing at least one phenol (or aromatic hydroxyl) group |
| WO2016004166A1 (en) * | 2014-07-02 | 2016-01-07 | Xavier University Of Louisiana | Boron-based prodrug strategy for increased bioavailability and lower-dosage requirements for drug molecules containing at least one phenol (or aromatic hydroxyl) group |
| WO2017086430A1 (en) * | 2015-11-20 | 2017-05-26 | 宇部興産株式会社 | Pharmaceutical composition for treatment or prophylaxis of nash |
| CN108348610A (en) * | 2015-11-20 | 2018-07-31 | 宇部兴产株式会社 | Medical composition for treating or preventing NASH |
| CN111032647A (en) * | 2017-09-04 | 2020-04-17 | 伊莱利利公司 | Lysophosphatidic acid receptor 1(LPAR1) inhibitor compounds |
| US11365185B2 (en) | 2017-09-04 | 2022-06-21 | Eli Lilly And Company | Lysophosphatidic acid receptor 1 (LPAR1) inhibitor compounds |
| AU2018323969B2 (en) * | 2017-09-04 | 2020-07-16 | Eli Lilly And Company | Lysophosphatidic acid receptor 1 (LPAR1) inhibitor compounds |
| EA039482B1 (en) * | 2017-09-04 | 2022-02-01 | Эли Лилли Энд Компани | Lysophosphatidic acid receptor 1 (lpar1) inhibitor compounds |
| KR102341064B1 (en) | 2017-09-04 | 2021-12-20 | 일라이 릴리 앤드 캄파니 | Lysophosphatidic acid receptor 1 (LPAR1) inhibitor compounds |
| CN111032647B (en) * | 2017-09-04 | 2023-05-02 | 伊莱利利公司 | Lysophosphatidic acid receptor 1 (LPAR 1) inhibitor compounds |
| JP2020532550A (en) * | 2017-09-04 | 2020-11-12 | イーライ リリー アンド カンパニー | Lysophosphatidic acid receptor 1 (LPAR1) inhibitor compound |
| KR20200035440A (en) * | 2017-09-04 | 2020-04-03 | 일라이 릴리 앤드 캄파니 | Lysophosphatidic acid receptor 1 (LPAR1) inhibitor compounds |
| WO2019046239A1 (en) * | 2017-09-04 | 2019-03-07 | Eli Lilly And Company | Lysophosphatidic acid receptor 1 (lpar1) inhibitor compounds |
| CN112055711A (en) * | 2017-12-19 | 2020-12-08 | 百时美施贵宝公司 | Cyclohexyl acid triazole azines as LPA antagonists |
| WO2019126094A1 (en) * | 2017-12-19 | 2019-06-27 | Bristol-Myers Squibb Company | Cyclohexyl acid triazole azoles as lpa antagonists |
| JP2021507898A (en) * | 2017-12-19 | 2021-02-25 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Pyrazole cyclohexylate as an LPA antagonist |
| JP2021506860A (en) * | 2017-12-19 | 2021-02-22 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Triazole acrylate cyclohexyl as an LPA antagonist |
| JP2021508686A (en) * | 2017-12-19 | 2021-03-11 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Triazole N-linked carbamoylcyclohexyl as an LPA antagonist |
| CN112521368A (en) * | 2017-12-19 | 2021-03-19 | 百时美施贵宝公司 | Triazole N-linked carbamoylcyclohexanoic acids as LPA antagonists |
| WO2019126090A1 (en) * | 2017-12-19 | 2019-06-27 | Bristol-Myers Squibb Company | Triazole n-linked carbamoyl cyclohexyl acids as lpa antagonists |
| US11008300B2 (en) | 2017-12-19 | 2021-05-18 | Bristol-Myers Squibb Company | Triazole n-linked carbamoyl cyclohexyl acids as LPA antagonists |
| JP7274486B6 (en) | 2017-12-19 | 2024-02-15 | ブリストル-マイヤーズ スクイブ カンパニー | Triazole N-linked carbamoylcyclohexylate as an LPA antagonist |
| CN112521368B (en) * | 2017-12-19 | 2023-12-05 | 百时美施贵宝公司 | Triazole N-linked carbamoyl cyclohexyl acid as LPA antagonists |
| CN111712492A (en) * | 2017-12-19 | 2020-09-25 | 百时美施贵宝公司 | Cyclohexaneacetic acid triazolazoles as LPA antagonists |
| US11697646B2 (en) | 2017-12-19 | 2023-07-11 | Bristol-Myers Squibb Company | Triazole N-linked carbamoyl cyclohexyl acids as LPA antagonists |
| US11180488B2 (en) | 2017-12-19 | 2021-11-23 | Bristol-Myers Squibb Company | Isoxazole o-linked carbamoyl cyclohexyl acids as LPA antagonists |
| JP7280881B2 (en) | 2017-12-19 | 2023-05-24 | ブリストル-マイヤーズ スクイブ カンパニー | Cyclohexylate triazole azoles as LPA antagonists |
| JP7274486B2 (en) | 2017-12-19 | 2023-05-16 | ブリストル-マイヤーズ スクイブ カンパニー | Triazole N-linked carbamoylcyclohexylates as LPA antagonists |
| US11447475B2 (en) | 2017-12-19 | 2022-09-20 | Bristol-Myers Squibb Company | Isoxazole N-linked carbamoyl cyclohexyl acids as LPA antagonists |
| US11261174B2 (en) | 2017-12-19 | 2022-03-01 | Bristol-Myers Squibb Company | Pyrazole O-linked carbamoyl cyclohexyl acids as LPA antagonists |
| US11261180B2 (en) | 2017-12-19 | 2022-03-01 | Bristol-Myers Squibb Company | Cyclohexyl acid isoxazole azoles as LPA antagonists |
| US11267800B2 (en) | 2017-12-19 | 2022-03-08 | Bristol-Myers Squibb Company | Cyclohexyl acid triazole azines as LPA antagonists |
| AU2018392321B2 (en) * | 2017-12-19 | 2022-03-10 | Bristol-Myers Squibb Company | Triazole N-linked carbamoyl cyclohexyl acids as LPA antagonists |
| IL275348B1 (en) * | 2017-12-19 | 2023-04-01 | Bristol Myers Squibb Co | Triazole n-linked carbamoyl cyclohexyl acids as lpa antagonists |
| US11312706B2 (en) | 2017-12-19 | 2022-04-26 | Bristol-Myers Squibb Company | Cyclohexyl acid pyrazole azines as LPA antagonists |
| US11319315B2 (en) | 2017-12-19 | 2022-05-03 | Bristol-Myers Squibb Company | Cyclohexyl acid triazole azoles as LPA antagonists |
| US11319309B2 (en) | 2017-12-19 | 2022-05-03 | Bristol-Myers Squibb Company | Cyclohexyl acid isoxazole azines as LPA antagonists |
| US11352345B2 (en) | 2017-12-19 | 2022-06-07 | Bristol-Myers Squibb Company | Cyclohexyl acid pyrazole azoles as LPA antagonists |
| JP7212047B2 (en) | 2017-12-19 | 2023-01-24 | ブリストル-マイヤーズ スクイブ カンパニー | Cyclohexylate Pyrazole Azoles as LPA Antagonists |
| EP4011875A1 (en) * | 2017-12-19 | 2022-06-15 | Bristol-Myers Squibb Company | Triazole n-linked carbamoyl cyclohexyl acids as lpa antagonists |
| US10662172B2 (en) | 2017-12-19 | 2020-05-26 | Bristol-Myers Squibb Company | Triazole N-linked carbamoyl cyclohexyl acids as LPA antagonists |
| JP7208240B2 (en) | 2017-12-19 | 2023-01-18 | ブリストル-マイヤーズ スクイブ カンパニー | Cyclohexylate triazole azines as LPA antagonists |
| US11384067B2 (en) | 2017-12-19 | 2022-07-12 | Bristol-Myers Squibb Company | Pyrazole n-linked carbamoyl cyclohexyl acids as LPA antagonists |
| JP2021507900A (en) * | 2017-12-19 | 2021-02-25 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Triazole cyclohexylate as an LPA antagonist |
| WO2019126093A1 (en) * | 2017-12-19 | 2019-06-27 | Bristol-Myers Squibb Company | Cyclohexyl acid triazole azines as lpa antagonists |
| WO2019246109A1 (en) | 2018-06-18 | 2019-12-26 | Epigen Biosciences, Inc. | Heterocyclic compounds useful in the treatment of disease |
| JP2021529733A (en) * | 2018-06-18 | 2021-11-04 | エピゲン バイオサイエンシズ, インコーポレイテッドEpigen Biosciences, Inc. | Heterocyclic compounds useful in the treatment of diseases |
| EP3807254A4 (en) * | 2018-06-18 | 2022-03-23 | Epigen Biosciences, Inc. | Heterocyclic compounds useful in the treatment of disease |
| CN111434653A (en) * | 2019-01-15 | 2020-07-21 | 武汉朗来科技发展有限公司 | Triazole compound and preparation method and application thereof |
| EP3912974A4 (en) * | 2019-01-15 | 2022-11-30 | Wuhan LL Science And Technology Development Co., Ltd. | Triazole compounds, and preparation method thereof and use |
| CN112654606A (en) * | 2019-01-15 | 2021-04-13 | 武汉朗来科技发展有限公司 | Triazole compound and preparation method and application thereof |
| CN112654606B (en) * | 2019-01-15 | 2023-11-07 | 武汉朗来科技发展有限公司 | Triazole compound as well as preparation method and application thereof |
| WO2020147740A1 (en) * | 2019-01-15 | 2020-07-23 | 武汉朗来科技发展有限公司 | Triazole compounds, and preparation method thereof and use |
| WO2020257135A1 (en) * | 2019-06-18 | 2020-12-24 | Bristol-Myers Squibb Company | Triazole carboxylic acids as lpa antagonists |
| JP7431961B2 (en) | 2019-11-15 | 2024-02-15 | ギリアード サイエンシーズ, インコーポレイテッド | Triazole carbamate pyridyl sulfonamides and their use as LPA receptor antagonists |
| WO2021097039A1 (en) * | 2019-11-15 | 2021-05-20 | Gilead Sciences, Inc. | Triazole carbamate pyridyl sulfonamides as lpa receptor antagonists and uses thereof |
| CN114728168B (en) * | 2019-11-15 | 2024-04-09 | 吉利德科学公司 | Triazole carbamate pyridylsulfonamides as LPA receptor antagonists and their use |
| TWI767410B (en) * | 2019-11-15 | 2022-06-11 | 美商基利科學股份有限公司 | Triazole carbamate pyridyl sulfonamides as lpa receptor antagonists and uses thereof |
| CN114728168A (en) * | 2019-11-15 | 2022-07-08 | 吉利德科学公司 | Triazolecarbamate pyridylsulfonamides as LPA receptor antagonists and their use |
| US11548871B2 (en) | 2019-11-15 | 2023-01-10 | Gilead Sciences, Inc. | Triazole carbamate pyridyl sulfonamides as LPA receptor antagonists and uses thereof |
| US11584738B2 (en) | 2020-06-03 | 2023-02-21 | Gilead Sciences, Inc. | LPA receptor antagonists and uses thereof |
| US11702407B2 (en) | 2020-06-03 | 2023-07-18 | Gilead Sciences, Inc. | LPA receptor antagonists and uses thereof |
| US11912686B2 (en) | 2020-06-03 | 2024-02-27 | Gilead Sciences, Inc. | LPA receptor antagonists and uses thereof |
| CN116669727A (en) * | 2020-08-11 | 2023-08-29 | 唯久生物有限公司 | Triazole-pyridinyl substituted azacyclohexylacetic acid compounds as LPA receptor antagonists |
| WO2022034568A1 (en) * | 2020-08-11 | 2022-02-17 | Viva Star Biosciences Limited | Triazole-pyridinyl substituted azacyclohexyl acetic acid compounds as lpa receptor antagonists |
| WO2022240879A1 (en) * | 2021-05-11 | 2022-11-17 | Gilead Sciences, Inc. | Lpa receptor antagonists and uses thereof |
| TWI818538B (en) * | 2021-05-11 | 2023-10-11 | 美商基利科學股份有限公司 | Lpa receptor antagonists and uses thereof |
| US11939318B2 (en) | 2021-12-08 | 2024-03-26 | Gilead Sciences, Inc. | LPA receptor antagonists and uses thereof |
| US11884627B2 (en) | 2022-02-25 | 2024-01-30 | Lhotse Bio, Inc. | Compounds and compositions for treating conditions associated with LPA receptor activity |
| WO2024022314A1 (en) * | 2022-07-25 | 2024-02-01 | 武汉人福创新药物研发中心有限公司 | Triazole compounds and use thereof as lpar1 antagonist |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2012138648A1 (en) | Compositions and methods for modulating lpa receptors | |
| AU2016256728B2 (en) | Thiadiazole analogs thereof and methods for treating smn-deficiency-related-conditions | |
| JP6130964B2 (en) | APJ receptor triazole agonist | |
| KR102368439B1 (en) | 1,4-disubstituted pyridazine analogs and methods for treating smn-deficiency-related conditions | |
| ES2525581T3 (en) | N-sulfonylbenzamide derivatives useful as voltage-dependent sodium channel inhibitors | |
| JP6452703B2 (en) | WNT pathway modulator | |
| TW201835072A (en) | Novel cyp11a1 inhibitors | |
| WO2012020820A1 (en) | Heteroaryl-pyrazole derivative | |
| CA2934010A1 (en) | N-acylpiperidine ether tropomyosin-related kinase inhibitors | |
| JP6670832B2 (en) | Novel dihydroquinoline pyrazolyl compounds as aldosterone synthase inhibitors | |
| JP6622300B2 (en) | Novel dihydroquinoline pyrazolyl compounds | |
| JP6669734B2 (en) | Pyrazolyl-3,4-didohydroquinolin-2-one aldosterone synthase inhibitors | |
| TW201910325A (en) | Indole derivatives and uses thereof | |
| WO2019087163A1 (en) | Polycyclic herg activators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12714181 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12714181 Country of ref document: EP Kind code of ref document: A1 |