WO2011070592A2 - Novel sugar derivatives - Google Patents
Novel sugar derivatives Download PDFInfo
- Publication number
- WO2011070592A2 WO2011070592A2 PCT/IN2010/000796 IN2010000796W WO2011070592A2 WO 2011070592 A2 WO2011070592 A2 WO 2011070592A2 IN 2010000796 W IN2010000796 W IN 2010000796W WO 2011070592 A2 WO2011070592 A2 WO 2011070592A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- aryl
- formula
- heteroaryl
- substituted
- Prior art date
Links
- 0 *N(*)*Cc1ccc(Cc2cc([C@@]([C@@](C3)O)O[C@](CO)[C@@]3O)ccc2Cl)cc1 Chemical compound *N(*)*Cc1ccc(Cc2cc([C@@]([C@@](C3)O)O[C@](CO)[C@@]3O)ccc2Cl)cc1 0.000 description 1
- DSQCKWXZFALMKC-ZQGJOIPISA-N CC(NS(NCCOc1ccc(Cc2cc([C@@H]([C@@H]([C@H]3O)O)O[C@H](CO)[C@H]3O)ccc2Cl)cc1)(=O)=O)=O Chemical compound CC(NS(NCCOc1ccc(Cc2cc([C@@H]([C@@H]([C@H]3O)O)O[C@H](CO)[C@H]3O)ccc2Cl)cc1)(=O)=O)=O DSQCKWXZFALMKC-ZQGJOIPISA-N 0.000 description 1
- WDDJGJUIWJXBPJ-UHFFFAOYSA-N CCP1(CCN(C)CC1)=O Chemical compound CCP1(CCN(C)CC1)=O WDDJGJUIWJXBPJ-UHFFFAOYSA-N 0.000 description 1
- CCBRCIPIHMSBQL-UHFFFAOYSA-N CN(CC1)CCP1(c1ccccc1)=O Chemical compound CN(CC1)CCP1(c1ccccc1)=O CCBRCIPIHMSBQL-UHFFFAOYSA-N 0.000 description 1
- FDUFPSGUEWXRNO-LDFCNSTISA-N CS(NCCOc1ccc(Cc2cc([C@@H]([C@@H]([C@H]3O)O)O[C@H](CN4CCCCC4)[C@H]3O)ccc2Cl)cc1)(=C)=[U] Chemical compound CS(NCCOc1ccc(Cc2cc([C@@H]([C@@H]([C@H]3O)O)O[C@H](CN4CCCCC4)[C@H]3O)ccc2Cl)cc1)(=C)=[U] FDUFPSGUEWXRNO-LDFCNSTISA-N 0.000 description 1
- IOTRLIDUQUIIRP-SEFGFODJSA-N CS(NCCOc1ccc(Cc2cc([C@@H]([C@@H]([C@H]3O)O)O[C@H](CNC(NC4CCCC4)=O)[C@H]3O)ccc2Cl)cc1)(=O)=O Chemical compound CS(NCCOc1ccc(Cc2cc([C@@H]([C@@H]([C@H]3O)O)O[C@H](CNC(NC4CCCC4)=O)[C@H]3O)ccc2Cl)cc1)(=O)=O IOTRLIDUQUIIRP-SEFGFODJSA-N 0.000 description 1
- XSOPRGCIXUTBND-WJMWBRGCSA-N NS(NCCOc1ccc(Cc(cc([C@@H]([C@@H]2O)O[C@H](CO)C[C@@H]2O)cc2)c2Cl)cc1)(=O)=O Chemical compound NS(NCCOc1ccc(Cc(cc([C@@H]([C@@H]2O)O[C@H](CO)C[C@@H]2O)cc2)c2Cl)cc1)(=O)=O XSOPRGCIXUTBND-WJMWBRGCSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/02—Acyclic radicals, not substituted by cyclic structures
- C07H15/04—Acyclic radicals, not substituted by cyclic structures attached to an oxygen atom of the saccharide radical
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/10—Oxygen atoms
Definitions
- the present invention relates to novel compounds of Formula I, their pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
- the invention also relates to the processes for the synthesis of novel compounds of Formula I, their pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
- the present invention also provides pharmaceutical compositions comprising novel compounds of Formula I and methods of treating or preventing one or more conditions or diseases that may be regulated or normalized via inhibition of Sodium Glucose Cotransporter-2 (SGLT-2).
- SGLT-2 Sodium Glucose Cotransporter-2
- the invention also relates to the use of compounds of Formula I, their pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, for the manufacture of a medicament for the prophylaxis, amelioration and/or treatment of conditions or diseases that may be regulated or normalized via inhibition of Sodium Glucose Cotransporter-2 (SGLT-2) and the related diseases, disorders and conditions, in a subject in need thereof.
- SGLT-2 Sodium Glucose Cotransporter-2
- Diabetes is a metabolic disorder which is rapidly emerging as a global health care problem that threatens to reach pandemic levels.
- the number of people with diabetes worldwide is expected to rise from 285 million in 2009 to 435 million by 2030. Diabetes results from deficiency in insulin because of impaired pancreatic ⁇ -cell function or from resistance to insulin in body, thus leading to abnormally high levels of blood glucose.
- Type 1 diabetes Diabetes which results from complete deficiency in insulin secretion is type 1 diabetes and the diabetes due to resistance to insulin activity together with an inadequate insulin secretion is type 2 diabetes.
- Type 2 diabetes Non insulin dependent diabetes
- An early defect in type 2 diabetes mellitus is insulin resistance which is a state of reduced responsiveness to circulating concentrations of insulin and is often present years before clinical diagnosis of diabetes.
- a key component of the pathophysiology of type 2 diabetes mellitus involves an impaired pancreatic ⁇ -cell function which eventually contributes to decreased insulin secretion in response to elevated plasma glucose. The ⁇ -cell compensates for insulin resistance by increasing the insulin secretion, eventually resulting in reduced ⁇ -cell mass. Consequently, blood glucose levels stay at abnormally high levels (hyperglycemia).
- Hyperglycemia is central to both the vascular consequences of diabetes and the progressive nature of the disease itself. Chronic hyperglycemia leads to decrease in insulin secretion and further to decrease in insulin sensitivity. As a result, the blood glucose concentration is increased, leading to diabetes, which is self-exacerbated. Chronic hyperglycemia has been shown to result in higher protein glycation, cell apoptosis and increased oxidative stress; leading to complications such as cardiovascular disease, stroke, nephropathy, retinopathy (leading to visual impairment or blindness), neuropathy, hypertension, dyslipidemia, premature atherosclerosis, diabetic foot ulcer and obesity. So, when a person suffers from diabetes, it becomes important to control the blood glucose level. Normalization of plasma glucose in type 2 diabetes patients improves insulin action and may offset the development of beta cell failure and diabetic complications in the advanced stages of the disease.
- Diabetes is basically treated by diet and exercise therapies. However, when sufficient relief is not obtained by these therapies, medicament is prescribed alongwith.
- Various anti-diabetic agents being currently used include biguanides (decrease glucose production in the liver and increase sensitivity to insulin), sulfonylureas and meglitinides (stimulate insulin production), a-glucosidase inhibitors (slow down starch absorption and glucose production) and thiazolidinediones (increase insulin sensitivity).
- DPP-IV dipeptidyl peptidase-IV
- GLP-1 glucagon like peptide- 1
- GIP gastric inhibitory peptide
- SGLTs sodium glucose co-transporters
- SGLT-1 is a low capacity, high-affinity transporter expressed in the gut (small intestine epithelium), heart, and kidney (S3 segment of the renal proximal tubule), whereas SGLT-2 (a 672 amino acid protein containing 14 membrane-spanning segments), is a low affinity, high capacity glucose transporter, located mainly in the SI segment of the proximal tubule of the kidney.
- SGLT-2 facilitates approximately 90% of glucose reabsorption and the rate of glucose filtration increases proportionally as the glycemic level increases.
- the inhibition of SGLT-2 should be highly selective, because non-selective inhibition leads to complications such as severe, sometimes fatal diarrhea, dehydration, peripheral insulin resistance, hypoglycemia in CNS and an impaired glucose uptake in the intestine.
- the first known non-selective SGLT-2 inhibitor was the natural product phlorizin
- C-glycoside derivatives have been disclosed, for example, in PCT publications WO2004013118, WO2005085265, WO2006008038, WO2006034489, WO2006037537, WO2006010557, WO2006089872, WO2006002912, WO2006054629, WO2006064033, WO20071361 16, WO2007000445, WO2007093610, WO2008069327, WO200802001 1, WO2008013321, WO2008013277, WO2008122014, WO2008116195, WO2008042688, WO2009026537 and WO2010022313, US patents US6515117B2, US6936590B2 and US7202350B2 and Japanese patent application JP2004359630.
- O-glycoside derivatives have been disclosed, for example, in PCT publications WO2002088157, WO2002064606, WO2003020737, WO2003000712, WO2004089966, WO2004058790, WO2004099230, WO2004087727, WO2005085267, WO2005095429, WO2005021566, WO200601 1469 and WO20071261 17 and US patents US6555519B2, US6683056B2, US6872706B2, US7056892B2, US7129381B2, US7189702B2, US7247616B2 and US7294618B2.
- the compounds shown below are the SGLT-2 inhibitors which have reached advanced stages of human clinical trials: Bristol-Myers Squibb's "Dapagliflozin” with Formula A and Mitsubishi Tanabe and Johnson & Johnson's "Canagliflozin” with Formula B.
- Various other compounds whose structures are not disclosed yet but are known to be in different phases of clinical trials are: Lexicon's Lx 421 1, Boehringer's BI 10773,
- the present invention relates to the novel compounds of Formula I,
- ring A represents aryl
- ring B represents either aryl or heteroaryl
- Y represents either O or S
- R 1 , R 2 , R 3 and R 4 are independently selected from hydrogen, halogen, Ci_i 2 alkyl, C 2 . ]2 alkenyl, C 2- i 2 alkynyl, C 2 _i 2 haloalkenyl, C 2 _i 2 haloalkynyl, Ci-i 2 alkoxy, Ci_ C]_i 2 alkylcarbonyl, Ci- 1 2 alkoxycarbonyl, C -2 ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2) n -cycloalkyl, cycloalkenyl, cycloalkynyl, -(CH 2 ) classroom-heterocyclyl, -(CH 2 ) n -aryl, -(CH 2 ) n -heteroary], - CN, -N0 2 , -NR 12 R 13 , -(CH2) n NR l2 R 13 ,
- E can be absent or is selected from CH 2 , O, S or NR i6 ;
- both R 5 and R 6 can not be hydrogen at the same time
- both R 5 and R 6 can not be alkyl at the same time
- R 5 and R 6 can not be a combination of hydrogen and alkyl at the same time
- R 6 when E and G are absent and R 5 is hydrogen then R 6 can not represent -S(0) d R a (e) when R 7 represents C 2- i 2 alkenyl, C 2- i 2 alkynyl, -CH 2 OH or -(CH 2 ) n R e , wherein n is not equal to zero; one of R 5 and R 6 represents -H or Ci -6 alkyl and the other represents -S(0)dR a , wherein d represents 1 or 2; then R a can not be C ⁇ . 6alkyl, C 2 . 6 alkenyl, C 2-6 alkynyl, aryl or heteroaryl;
- R 8 , R 9 , R 10 , R 12 , R 13 , R 14 and R 15 are independently selected from hydrogen, halogen, Cj . nalkyl, C 2- i 2 alkenyl, C 2 .i alkynyl, Ci_i 2 alkoxycarbonyl, C 3 .
- R n is selected from hydrogen, halogen, Ci -]2 alkyl, C 2- i 2 alkenyl, C 2 _i 2 alkynyl, Ci_ i 2 haloalkyl, C 2 _i 2 haloalkenyl, C 2 _i 2 haloalkynyl, Ci_
- i 2 alkyl C 2- i 2 alkenyl, C 2 .i 2 alkynyl, C 3-20 cycloalkyl, heterocyclyl, aryl, heteroaryl, , -CN, - N0 2 orNH 2 ;
- R a , R b and R c are independently selected from hydrogen, halogen, C
- n 0, 1, 2, 3, 4 or 5;
- d is 1 or 2;
- n 1, 2, 3, 4 or 5.
- a further aspect of the present invention provides processes for the preparation of the novel compounds of Formula I, their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
- compositions containing compounds of Formula I, their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof in combination with one or more pharmaceutically acceptable carrier(s), adjuvants and vehicles.
- Another aspect of the present invention is the use of the compounds of Formula I, their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, for the prophylaxis, amelioration and/or treatment of one or more condition(s)/disease(s)/ disorder(s), in a subject in need thereof.
- Still another aspect of the present invention is the use of the compounds of Formula I, their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, for the prophylaxis, amelioration and/or treatment of one or more condition(s)/disease(s)/ disorder(s) that may be regulated or normalized via inhibition of SGLT-2.
- Yet another aspect of the invention is to provide methods of using the compounds of Formula I of the present invention or compositions comprising the compounds of Formula I for the prophylaxis, amelioration and/or treatment of disease(s)/ disorder(s) involving SGLT-2 inhibition which comprises administering to a subject in need thereof the compounds of Formula I or compositions comprising a pharmaceutically effective amount of the compounds of Formula I.
- a further aspect of the present invention is the use of a compound of Formula I for the manufacture of a medicament for the prophylaxis, amelioration and/or treatment of one or more condition(s)/disease(s)/ disorder(s) involving SGLT-2 inhibition in a subject in need thereof.
- the present invention also encompasses prodrugs and active metabolites of the compounds of the Formula I. 0796
- the present invention relates to the novel compounds of Formula I,
- ring A represents aryl
- ring B represents either aryl or heteroaryl
- Y represents either O or S
- R 1 , R 2 , R 3 and R 4 are independently selected from hydrogen, halogen, Ci_i 2 alkyl, C 2- i 2 alkenyl, C 2 _i 2 alkynyl, Ci-i 2 haloalkyl, C 2 -i 2 haloalkenyl, C 2 .i 2 haloalkynyl, Ci_i 2 alkoxy, C ⁇ . n haloalkoxy, Ci -6 alkoxyCi- 6 alkyl, C]- 6 alkoxyCi. 6 alkoxyCi.
- E can be absent or is selected from CH 2 , O, S or NR 16 ;
- both R 5 and R 6 can not be hydrogen at the same time
- both R 5 and R 6 can not be alkyl at the same time
- R 5 and R 6 can not be a combination of hydrogen and alkyl at the same time
- R 7 represents Cu 2 alkyl, C 2- i 2 alkenyl, C 2- i 2 alkynyl, -CH 2 OH or -(CH 2 ) n R e , wherein n is not equal to zero; one of R 5 and R 6 represents -H or C 1-6 alkyl and the other represents -S(0) d R a , wherein d represents 1 or 2; then R a can not be Ci. 6alkyl, C 2- alkenyl, C 2-6 alkynyl, aryl or heteroaryl;
- R 8 , R 9 , R 10 , R 12 , R 13 , R 14 and R 15 are independently selected from hydrogen, halogen, Q. i 2 alkyl, C 2- i 2 alkenyl, C 2- i 2 alkynyl, Ci -)2 alkylcarbonyl, Ci -)2 alkoxycarbonyl, C 3- 2 ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH 2 ) n -cycloalkyl, -(CH 2 ) n -heterocyclyl, - (CH 2 ) n -aryl, -(CH 2 ) n -heteroaryl, -CN, -N0 2, -NR a R b , -(CH 2 ) n NR a R b , -N 3 , -NCS, - (CH 2 ) complicatN 3 , -(CH 2 ) n NCS, -CR
- R 1 1 is selected from hydrogen, halogen, Ci-i 2 alkyl, C 2 .i 2 alkenyl, C 2 -i 2 alkynyl, C
- R a , R b and R c are independently selected from hydrogen, halogen, Ci-i 2 alkyl, C 2-12 alkenyl, C 2- i 2 alkynyl, Ci -12 alkoxy, Ci. 6 alkoxyCi -6 alkyl, Ci_i 2 alkylcarbonyl, Ci_i 2 alkoxycarbonyl, C 3-20 cycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH 2 ) n -cycloalkyl, -(CH 2 ) n -heterocyclyl, -(-CH 2 ) n -aryl, -(CH 2 ) n -heteroaryl, -CN, -N0 2 , -N 3 , -NCS, -NR 8 R 9 , -(CH 2 ) n NR 8 R 9 , - (CH 2 ) 11 N 3 , -(CH 2 ) n NCS, -
- n 0, 1, 2, 3, 4 or 5;
- d is 1 or 2;
- n 1, 2, 3, 4 or 5.
- One embodiment of the present invention provides compounds of Formula la, wherein
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , U, V, W, E, G, ring A and ring B are as defined herein; their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
- Another embodiment of the present invention provides compounds of Formula lb, wherein
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , E, G, ring A and ring B are as defined herein; their
- Another embodiment of the present invention provides compounds of Fonnula Ic, wherein
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , E and G are as defined herein; their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
- Another embodiment of the present invention provides compounds of Formula Id, wherein
- R 5 , R 6 , R 7 , E and G are as defined herein; their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
- E is selected from O or CH 2 .
- G is C 1-12 alkylene which is unsubstituted or substituted at any available position by one or more substituents selected from R n .
- R 7 is selected from the group consisting of -OCH3, -CH 2 OH, - CH 2 OCH 2 CF 3 , -CH 2 OCOCH 3 , -CH 2 OCOC 2 H 5 , -CH 2 OCOC 3 H 7 , -CH 2 OCOC 4 H 9 , - CH 2 OCO(CH 2 ) 5 CH 3 , -CH 2 OCO(CH 2 ) 7 CH 3 , -CH 2 OCO(CH 2 ) 10 CH 3 ,
- R 5 and R 6 are independently selected from the group consisting of -H, -CH 3 , -S0 2 CH 3 ,
- alkyl refers to a straight or branched chain aliphatic hydrocarbon chain, having from 1 to 12 carbon atoms.
- alkyl include, but are not limited to methyl, ethyl, n-propyl, isoprppyl, n-butyl, n-pentyl, t-butyl and the like.
- These groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
- substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
- alkenyl refers to a straight or branched chain aliphatic hydrocarbon group containing at least one carbon- carbon double bond, having from 2 to 12 carbon atoms.
- alkenyl include, but are not limited to ethenyl, 1-propenyl, 2-propenyl, iso-propenyl, 1 -butenyl, 2-butenyl, and the like.
- alkynyl refers to a straight or branched chain aliphatic hydrocarbon group containing at least one carbon- carbon triple bond, having from 2 to 12 carbon atoms.
- alkynyl examples include, but are not limited to ethynyl, propynyl, and butynyl. These groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
- alkylene refers to a divalent straight or branched chain aliphatic hydrocarbon group, having from 1 to 12 carbon atoms.
- alkylene include, but are not limited to methylene, ethylene, isopropylene, n-butylene, 1 ,1 - dimethylethylene and the like. These groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
- alkenylene refers to a divalent straight or branched chain aliphatic hydrocarbon group containing at least one carbon-carbon double bond, having from 2 to 12 carbon atoms.
- alkenyl include, but are not limited to ethenylene, 1 -propenylene, 2-propenylene, iso-propenylene, 1 -butenylene, 2-butenylene, and the like.
- These groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
- substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
- alkynylene refers to a divalent straight or branched chain aliphatic hydrocarbon group containing at least one carbon-carbon triple bond, having from 2 to 12 carbon atoms.
- alkynyl include, but are not limited to ethynylene, propynylene, and butynylene. These groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
- alkoxy refers to an above defined alkyl group attached via an oxygen linkage to the rest of the molecule. Non-limiting examples of such groups include -OCH 3 , -OC 2 3 ⁇ 4 and the like.
- alkoxyalkyl refers to an above defined alkyl group, in which one or more hydrogen atoms are replaced by alkoxy group as defined herein. Non-limiting examples include -CH 2 OCH 3 , -CH 2 OC 2 H 5 , -CH 2 CH 2 OC 2 H 5 and the like.
- alkoxyalkoxyalkyl refers to an above defined alkoxyalkyl group, in which one or more hydrogen atoms are replaced by above defined alkoxy group.
- Non- limiting examples include -CH 2 OCH 2 OCH 3 , -CH 2 OCH 2 CH 2 OC 2 H 5 , CH 2 CH 2 OCH 2 OC 2 H 5 and the like.
- alkyl carbonyl refers to an above defined alkyl group attached via a carbonyl linkage to the rest of the molecule.
- Non-limiting examples of such groups include -C(0)CH 3 , -C(0)C 2 H 5 and the like.
- alkoxycarbonyl refers to an above defined alkoxy group attached via a carbonyl linkage to the rest of the molecule.
- Non-limiting examples of such groups include -C(0)-0 CH 3 , -C(0)-OC 2 H 5 , and the like.
- halogen refers to F, CI, Br or I.
- haloalkyl refers to an above-defined “alkyl” group, which is substituted with one or more "halogen” groups, as defined herein, at any one or more of the 1 to 12 carbon atoms of the alkyl group.
- Representative examples of haloalkyl include, but are not limited to, chloromethyl, fluoromethyl, trifluoromethyl, trichloromethyl, difluoroethyl, trifluoroethyl, dichloroethyl, and the like.
- haloalkenyl refers to an above-defined “alkenyl” group, which is substituted with one or more "halogen” groups, as defined herein, at any one or more of the carbon atoms of the alkenyl group.
- Representative examples of haloalkenyl include, but are not limited to, chloroethenyl, 2-fluroethenyl, triflurobutenyl, dichloropropenyl and the like.
- haloalkynyl refers to an above-defined “alkynyl” group, which is substituted with one or more "halogen” groups, as defined herein, at any one or more of the carbon atoms of the alkynyl group.
- Representative examples of haloalkynyl include, but are not limited to, 2-fluroethynyl, triflurobutynyl, dichloropropynyl and the like.
- haloalkoxy refers to an above defined “haloalkyl” group, appended to the parent molecular moiety through an oxygen atom.
- cycloalkyl refers to cyclic alkyl groups consisting of 3 to 20 carbon atoms having a single cyclic ring or multiple condensed rings, for example, fused or spiro systems, which may be partially unsaturated, unless otherwise constrained by the definition.
- Such cycloalkyl groups include, by way of example, single ring structures, for example, cyclopropyl, cyclobutyl, cyclopentenyl, cyclohexyl, cyclooctyl, and the like, or multiple ring structures, for example, adamantyl, and bicyclo[2.2.1] heptane, or cyclic alkyl groups to which is fused an aryl group, for example, indane and the like.
- Cycloalkyl groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
- aryl refers to a mono- or poly- carbocyclic aromatic group, for example phenyl or naphthyl ring and the like optionally substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, alkyl, alkenyl, alkynyl, cycloalkyl, alkoxy, acyl, amino, aryloxy, CF 3 , COOR d (wherein R d can be hydrogen, alkyl, alkenyl, cycloalkyl, aralkyl, heterocyclylalkyl or heteroarylalkyl), cyano, nitro, carboxy, heterocyclyl, heteroaryl, heterocyclylalkyl or heteroarylalkyl.
- the aryl group may optionally be fused with cycloalkyl group, heteroaryl group, heterocyclyl group or another aryl group.
- the fused group may be further substituted at any available position with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, amino, nitro, cyano, carboxyalkyl, azido, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl, acyloxy, aryl or heterocyclyl, heteroaryl.
- aryloxy refers to an above defined aryl group attached via an oxygen linkage to the rest of the molecule, for example -OPh and the like.
- heteroaryl refers to an aromatic monocyclic or polycyclic ring structure, containing one or more heteroatoms independently selected from N, O, S or P.
- Heteroaryl also includes, but is not limited to, bicyclic or tricyclic rings, wherein the heteroaryl ring is fused to one or two rings independently selected from the group consisting of an aryl ring, a cycloalkyl ring, a heterocyclyl ring and another monocyclic heteroaryl ring.
- heteroaryl groups include, but not limited to, oxazolyl, imidazolyl, pyrrolyl, 1 ,2,3-triazolyl, 1,2,4-triazolyl, tetrazolyl, thiazolyl, ox_adiazolyl, benzoimidazolyl, thiadiazolyl, pyridinyl, pyridazinyl, pyrimidinyl, thienyl, isoxazolyl, triazinyl, furanyl, benzofuranyl, indolyl, benzothiazolyl, benzoxazolyl, imidazo[l,2-a]pyrimidine, imidazo[l ,2-a]pyrazine, and the like.
- the bicyclic or tricyclic heteroaryl rings can be attached either through the heteroaryl group itself or the aryl, cycloalkyl or heterocyclyl group to which it is fused.
- the heteroaryl group may be further substituted at any available position with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, amino, nitro, cyano, carboxyalkyl, azido, alkenyl, alkynyl, alkoxy, cycloalkyl, cycloalkynyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
- heterocyclyl refers to a non-aromatic monocyclic or polycyclic cycloalkyl group, for example, fused or spiro systems fully or partially unsaturated, containing one or more heteroatom(s) independently selected from N, O, S or P.
- the nitrogen, sulphur and phosphorus heteroatoms may optionally be oxidized.
- the nitrogen atoms may optionally be quaternerized.
- the heterocyclyl ring may be fused with another cycloalkyl, aryl, heterocyclyl or heteroaryl ring and are optionally benzofused or fused heteroaryl of 5-6 ring members and/or are optionally substituted wherein the substituents are selected from but not limited to halogen, hydroxy, alkyl, alkenyl, alkynyl, cycloalkyl, acyl, carboxy, aryl, alkoxy, aralkyl, cyano, nitro, amino, heterocyclyl, or heteroaryl.
- heterocyclyl groups include but are not limited to, morpholinyl, oxazolidinyl, tetrahydrofuranyl, dihydrofuranyl, dihydropyridinyl, dihydroisooxazolyl, dihydrobenzofuryl, azabicyclohexyl, dihydroindonyl, piperidinyl or piperazinyl.
- the fused group may be further substituted at any available position with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, amino, nitro, cyano, carboxyalkyl, azido, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
- substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, amino, nitro, cyano, carboxyalkyl, azido, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
- hydroxy refers to the group -OH.
- nitrogen, sulphur and phosphorus heteroatom can optionally be quaternerized or oxidized wherever possible.
- Protecting Group refers to a group which is in a modified form to preclude undesired side reactions at the protected site.
- protecting group may be used with groups, for example, hydroxy, amino, carboxy and examples of such groups are found in T.W. Greene, et al. "Protecting Groups in Organic Synthesis " 3 ld Ed, Wiley, New York, which is incorporated herein by reference.
- the species of the carboxylic protecting groups, amino protecting groups or hydroxy protecting groups employed are not critical, as long as the derivatised moieties/moiety is/are stable to conditions of subsequent reactions and can be removed without disrupting the remainder of the molecule.
- Suitable hydroxy and amino protecting groups include but are not limited to trimethylsilyl, triethylsilyl, o-nitrobenzyloxycarbonyl, p- nitrobenzyloxycarbonyl, f-butyldiphenylsilyl, i-butyldimethylsilyl, acetyl, trifluoroacetyl, benzyloxycarbonyl (CBz), t-butoxycarbonyl (Boc), 9-fluorenylnethylenoxycarbonyl (Fmoc), 2,2,2-trichloroethyloxycarbonyl, allyloxycarbonyl and the like.
- carboxy protecting groups examples include benzhydryl, o-nitrobenzyl, -nitrobenzyl, 2- naphthylmethyl, allyl, 2-chloroallyl, benzyl, 2,2,2- trichloroethyl, trimethylsilyl, t- butyldimethylsilyl, t-butyldiphenylsilyl, 2-(trimethylsilyl)ethyl, phenacyl, p- methoxybenzyl, acetonyl,/?-methoxyphenyl, 4-pyridylmethyl, -butyl and the like.
- Subject includes humans, non-human mammals (e.g., dogs, cats, rabbits, cattle, horses, sheep and the like) or non-mammals (e.g., birds and the like).
- non-human mammals e.g., dogs, cats, rabbits, cattle, horses, sheep and the like
- non-mammals e.g., birds and the like
- terapéuticaally effective amount means the amount of a compound that, when administered to a subject for treating a disease, is sufficient to effect such treatment for the disease.
- the “therapeutically effective amount” will vary depending on the compound, the disease and its severity, weight, physical condition and responsiveness of the subject to be treated, among other factors. -.
- a “pharmaceutically acceptable salt” refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- a “pharmaceutically acceptable salt” also encompasses any compound according to the present invention that is utilized in the form of a salt thereof.
- Asymmetric centres may exist in the compounds of the present invention.
- the compounds of Formula I may have one or more stereogenic centres and so can exhibit optical isomerism. All such isomers including enantiomers, diastereomers, and epimers are included within the scope of this invention.
- the invention includes such compounds as single isomers (R and /or S) and as mixtures, including racemates. If desired, racemic mixtures of the compounds may be separated so that the individual enantiomers are isolated.
- the separation may be carried out by methods well known in the art, such as the coupling of a racemic mixture of compounds to an enantiomerically pure compound to form a diastereomeric mixture, followed by separation of the individual diastereomers by standard methods, such as fractional crystallization or chromatography.
- Starting materials of particular stereochemistry may either be commercially available or may be made by the methods described herein and resolved by techniques well known in the art.
- the independent syntheses of these diastereomers or their chromatographic separations may be achieved as known in the art by appropriate modifications.
- Certain compounds according to Formula I can also exist as tautomers, which have different points of attachment of hydrogen accompanied by one or more double bond shifts. These tautomers, either separately or as mixtures, are also considered to be within the scope of the invention.
- Certain compounds according to Formula I may also exist as polymorphs.
- the present invention also encompasses geometrical isomers of compounds of 5 Formula I and the mixtures thereof.
- the geometrical isomers may exist in E or Z; Syn or anti configurations. These geometrical isomers, either separately or as mixtures, are also considered to be within the scope of the invention.
- Particularly useful examples of the present invention include but are not limited to the compounds selected from Table 1, including their pharmaceutically acceptable 10 derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof:
- the compounds disclosed herein may be prepared by techniques well known in the art and familiar to the skilled organic chemist.
- the compounds of the present invention may be prepared by the following reaction sequences as depicted in for example Scheme No 1 to 5. All of the starting materials are either commercially available or can be prepared by procedures that would be well known to one of ordinary skill in organic chemistry.
- Lg is used to denote an appropriate leaving group and as such may vary in nature depending on the exact reaction conditions employed. Some typical leaving groups may be fluoro, chloro, bromo, iodo, tosyl, mesyl, trifluoromethanesulfonyl and the like, but these should not be construed as limiting as many other leaving groups are also well known to those skilled in the art.
- the compounds of the Formula I can be prepared from the compounds of Formula II following the steps provided in Scheme 2 or Scheme 3.
- the compounds of the Formula II can be coupled with compounds of Formula III to obtain compounds of Formula IV.
- the compounds of Formula III contain a N- protection group (PG) such as but not limited to ter -butoxycarbonyl, Fluorenyloxycarbonyl, Allyloxycarbonyl or Benzyloxycarbonyl etc. and a leaving group (Lg) for example but not limited to a halogen, triflate, mesylate, tosylate or boronate and the like.
- PG N- protection group
- Lg leaving group
- the procedures to couple Formula II with Formula III include but not limited to, mitsunobu reaction involving reagents such as diethylazadicaorboxylate, diisopropylazadicarboxylate in presence of triphenylphosphene in a suitable solvent such as but not limited to THF, Dioxane, DCM and the like or catalytic reactions involving Pd, Cu etc metal derivatives in presence or absence of a ligand or procedures , involving treatment of Formula II with Formula III in presence of a base for example but not limited to NaH, KH, Cs 2 C0 , K 2 C0 3 , NEt 3 , pyridine and the like in a suitable solvent such as THF, Dioxane, DMF, DMSO, acetone, dichloroethane, DCM or combination there of.
- a suitable solvent such as but not limited to THF, Dioxane, DMF, DMSO, acetone, dichloroethane, DCM or combination
- the protection group from the compounds of Formula IV can be released by methods known to a person skilled in the art.
- Such deprotection methods include but not limited to, treatment with acids such as trifluoro acetic acid, HCl, H2SO 4 , HBr, HI etc.in a suitable solvent like DCM, Dichloroethane, diethylether, diisopropylether, THF, dioxane, actonitrile etc. or combination there of.
- Another type of deprotection methods include catalytic reductive methods involving Pd/C in presence of hydrogen in a suitable solvent such as but not limited to ethylacetate, methanol, acetic acid and the like or combination thereof.
- the compounds of Formula IV after deprotection can be converted to Formula I by treatement with compounds of Formula R 6 -Lg in presence of bases such as, but not limited to triethylamine, N-ethyldiisopropylamine, pyridine and the like in suitable solvents such as DCM, dichloroethane, THF, acetonitrile or combination thereof.
- bases such as, but not limited to triethylamine, N-ethyldiisopropylamine, pyridine and the like in suitable solvents such as DCM, dichloroethane, THF, acetonitrile or combination thereof.
- the compounds of the Formula II, wherein Q represents particularly, - Ci-i2alkyl-OH can be reacted with reagents such as but not limited to, methanesulfonylchloride, p-toluenesulfonylchloride, trifuorosulfonicanhydride, thionylchloride, phosphorousoxychloride, carbontetrachloride, borontribromide, phosphoroustribromide, carbontetrabromide, iodine etc.
- reagents such as but not limited to, methanesulfonylchloride, p-toluenesulfonylchloride, trifuorosulfonicanhydride, thionylchloride, phosphorousoxychloride, carbontetrachloride, borontribromide, phosphoroustribromide, carbontetrabromide, iodine etc.
- the compounds of Formula V can be converted to compounds of Formula VI by treating with sodiumazide in solvents such as but not limited to DMF, DMSO etc.
- the compounds of Formula VI can be converted to compounds of Formula VII by methods known to a person skilled in the art.
- Such methods include but not limited to treatment with triphenylphosphene in solvent like THF in presence of water or reduction in hydrogen atmosphere in presence of Pd/C to afford compounds of Formula VII.
- the compounds of Formula VII can be converted to Formula I by treatment with compounds of Formula R 6 -Lg in presence of bases such as, but not limited to triethylamine, JV-ethyldiisopropylamine, pyridine etc. in suitable solvents like DCM, dichloroethane, THF, acetonitrile or combination thereof.
- the compounds of the Formula II can be prepared from Formula VIII and compounds of Formula IX by following Scheme 4.
- Formula IX can be prepared by following the procedure given in NUCLEOSIDE, NUCLOETIDES & NUCLEIC ACIDS, 20(4-7), 649-652 (2001).
- the compounds of Formula VIII can be reacted with metalated species generated by treating Formula IX (prepared according to the procedure given in US20070049537) with reagents such as but not limited to, "BuLi, 'BuLi or 'PrMgCl in presence or absence of LiCl etc. in a suitable solvent such as but not limited to, THF, diiethylether or combination thereof to obtain the compounds of Formula X.
- the compounds of Formula X can be converted to the compounds of Formula II by deprotection methods kown to a person skilled in the art. These deprotection methods include but not limited to, treatment with acids such as trifluoro acetic acid, HC1, H 2 S0 4, HBr, HI etc.in a suitable solvent like DCM, Dichloroethane, diethylether, diisopropylether, THF, dioxane, actonitrile etc. or combination there of. eme 5
- acids such as trifluoro acetic acid, HC1, H 2 S0 4, HBr, HI etc.in a suitable solvent like DCM, Dichloroethane, diethylether, diisopropylether, THF, dioxane, actonitrile etc. or combination there of. eme 5
- Q-PG is -OPG, -SPG, -N(PG) 16 , -C ⁇ alkyl-OPG
- Q-PG is -OPG, -SPG, -N(PG)R 16 , -C-,. 12 alkyl-OPG
- Formula XI can be prepared by following the procedure given in US20070049537.
- Formula XI can be reacted with metalated species generated by treating Formula IX (prepared according to the procedure given in US20070049537) with reagents such as but not limited to, "BuLi, 'BuLi or 'PrMgCl in presence or absence of LiCl and the like in a suitable solvent such as but not limited to, THF, diiethylether or combination thereof.
- a suitable acetal formation reaction for example but not limited to, treatment with methanesulfonicacid in presence of methanol to obtain compounds of Formula XII.
- the compounds of Formula XII can be reduced by treating with triethylsilane in presence of borotri fluoride etherate in acetonitrile to furnish the compounds of Formula XIII.
- the compounds of Formula XIII can be converted to the compounds of Formula II by deprotection methods kown to a person skilled in the art. These deprotection methods include but not limited to, treatment with acids such as trifluoro acetic acid, HC1, H 2 S0 4j HBr, HI and the like in a suitable solvent like DCM, Dichloroethane, diethylether, diisopropylether, THF, dioxane, actonitrile and the like or combination there of.
- compounds of Formula XIII can be converted to compounds of Formula II by deprotection reactions involving reagents such as but not limited to tetrabutylammoniumfluoride or Hydrogenfluoride in solvents such as DCM, dichloroethane, THF, dioxane or pyridine and the like.
- reagents such as but not limited to tetrabutylammoniumfluoride or Hydrogenfluoride in solvents such as DCM, dichloroethane, THF, dioxane or pyridine and the like.
- references to the compounds of structural Formula I are meant to also include the pharmaceutically acceptable salts, and also salts that are not pharmaceutically acceptable when they are used as precursors to the free compounds or their pharmaceutically acceptable salts or in other synthetic manipulations.
- the compounds of the present invention may be administered in the form of a pharmaceutically acceptable salt.
- pharmaceutically acceptable salt refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids. The salts may be prepared during the final isolation and purification of the compounds or separately by making basic or acidic addition salts.
- Representative salts of basic compounds of the present invention can be prepared by reacting free base form of the compound with a suitable acid, including, but not limited to acetate, trifluoroacetate, adipate, citrate, aspartate, benzoate, benzenesulphonate, bisulfate, besylate, butyrate, camphorsulphonate, difluconate, hemisulfate, heptanoate, formate, fumarate, lactate, maleate, methanesulfonate, naphthylsulfonate, nicotinate, oxalate, picrate, pivalate, succinate, tartrate, tirchloracetat, glutamate, >-toluenesulphonate, hydrochloric, hydrobromic, sulphuric, phosphoric and the like.
- a suitable acid including, but not limited to acetate, trifluoroacetate, adipate, citrate, aspartate, benzoate, benz
- Representative salts of acidic compounds of the present invention can be prepared by reacting free acid form of the compound with a suitable base, including, but not limited to ammonium, calcium, magnesium, potassium, sodium salts, salts of primary, secondary and tertiary amines, substituted amines including naturally occurring ones e.g., arginine, betaine, caffeine, choline, glucamine, glucosamine, histidine, lysine, morpholine, piperazine, piperidine, purine, triethylamine and the like.
- a suitable base including, but not limited to ammonium, calcium, magnesium, potassium, sodium salts, salts of primary, secondary and tertiary amines, substituted amines including naturally occurring ones e.g., arginine, betaine, caffeine, choline, glucamine, glucosamine, histidine, lysine, morpholine, piperazine, piperidine, purine, triethylamine and
- solvates refer to solvates with water (i.e., hydrates) or pharmaceutically acceptable solvents, for example, ethanol and the like.
- prodrugs of the compounds of the present invention which upon in-vivo administration undergo cleavage by metabolic processes before becoming active pharmacological substances.
- prodrugs are derivatives of functional group of a compound of the invention which are readily convertible in vivo into the compound of the invention.
- Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in “Targeted prodrug design to optimize drug delivery", AAPS PharmaSci (2000), 2(1), E6.
- the invention also encompasses active "metabolites" of the compound of the present invention.
- An active metabolite is an active derivative of a SGLT-2 inhibitor produced when the SGLT-2 inhibitor is metabolized.
- polymorphs of a compound of general Formula I forming part of this invention may be prepared by crystallization of a compound of Formula I under different conditions. For example, by using different solvents commonly used or their mixtures for recrystallization; crystallizations at different temperatures; various modes of cooling, ranging from very fast to very slow cooling during crystallizations, heating or melting the compound followed by gradual or fast cooling may also obtain polymorphs. The presence of polymorphs may be determined by solid probe NMR spectroscopy, IR spectroscopy, differential scanning calorimetry, powder X-ray diffraction or such other techniques.
- the present invention includes all pharmaceutically acceptable isotopically- labeled compounds of Formula I wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes suitable for inclusion in the compounds of the present invention comprises isotopes of hydrogen, such as 2 H and 3 H, carbon, such as "C, 13 C and l4 C, chlorine, such as 36 C1, fluorine, such as F, iodine, such as I and I, nitrogen, such as N and N, oxygen, such as O,
- Isotopically- labeled compounds of Formula I can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying examples and schemes using an appropriate isotopically-labeled reagent in place of the non-labeled reagent previously employed.
- the present invention also includes all the intermediate complexes of the compounds of Formula I, which are active by themselves or can be readily converted to compounds having inhibitory effect on sodium-dependent glucose cotransporter (SGLT), preferably SGLT-2.
- SGLT sodium-dependent glucose cotransporter
- the present invention also provides pharmaceutical compositions, comprising compounds of general Formula I or their pharmaceutically acceptable analogs, derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof together with one or more pharmaceutically acceptable carriers comprising excipients and auxiliaries, which facilitate processing of the active compound into preparations, which can be used pharmaceutically.
- compositions may be in the forms normally employed, such as tablets, capsules, powders, syrups, solutions, suspensions, emulsions, pills, granules, suppositories, pellets, depot formulations and the like, may contain flavourants, sweeteners etc in suitable solid or liquid carriers or diluents, or in suitable sterile media to form injectable solutions or suspensions.
- Such compositions typically contain from 0.1 to 99.9 % by weight of active compound, the remainder of the composition being pharmaceutically acceptable carriers, diluents or solvents.
- compositions of the present invention can be manufactured by the processes well known in the art, for example, by means of conventional mixing, dissolving, dry granulation, wet granulation, dragee-making, levigating, emulsifying, encapsulating, entrapping, lyophilizing processes or spray drying.
- the compounds or the pharmaceutical compositions comprising such compounds of the present invention may be administered in the form of any pharmaceutical formulation.
- the pharmaceutical formulation will depend upon the nature of the active compound and its route of administration.
- any route of administration may be used, for example oral, buccal, pulmonary, topical, parenteral (including subcutaneous, intramuscular and intravenous), transdermal, ocular (ophthalmic), by inhalation, intranasal, transmucosal, implant or rectal administration.
- parenteral including subcutaneous, intramuscular and intravenous
- transdermal ocular
- ocular ophthalmic
- the compounds of the present invention are administered orally, parenterally or topically.
- the amount of the novel compounds having the Formula I according to the present invention to be incorporated into the pharmaceutical compositions of the present invention can vary over a wide range depending on known factors such as, for example, the disorder to be treated, the severity of the disorder, the patient's body weight, the dosage form, the chosen route of administration and the number of administration per day.
- the amount of the compound of Formula I in the pharmaceutical compositions of the present invention will range from approximately 0.01 mg to about 5000 mg.
- the daily dose of composition comprising the novel compounds having the Formula I is in the range of about 0.01 mg/kg to about 100 mg/kg based on the body weight of the subject in need thereof which may be administered as a single or multiple doses.
- novel compounds having the Formula I according to the present invention are particularly useful for the treatment of disease(s) or disorder(s), which are chronic or acute in nature, which favorably respond to or are alleviated by the novel compounds having the Formula I or compositions comprising them.
- the compositions comprising the novel compounds having the Formula I are useful prophylactically or therapeutically depending upon the pathological condition intended to be prevented or treated respectively.
- compounds of the present invention are useful in the prophylaxis, amelioration and/or treatment of one or more condition(s)/disease(s)/ disorder(s), in a subject in need thereof.
- compounds of the present invention are useful in the prophylaxis, amelioration and/or treatment of one or more condition(s)/disease(s)/ disorder(s), which may be regulated or normalized via inhibition of Sodium Glucose Cotransporters (SGLT).
- SGLT Sodium Glucose Cotransporters
- the compounds of the present invention possess activity as selective inhibitors of SGLT-2 and are therefore useful for the prophylaxis, amelioration and/or treatment of variety of diseases, disorders and conditions, including, but not limited to, diabetes (including Type I and Type II), Metabolic Syndrome or 'Syndrome X' including impaired glucose tolerance, insulin resistance, metabolic acidosis or ketosis, disorders of food intake, satiety disorders, obesity, hyperinsulinemia, dyslipidemia (including hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels, high LDL levels), hypertension associated with metabolic disorders, congestive heart failure, edema, hyperuricemia, gout, wound healing and tissue ischemia.
- diabetes including Type I and Type II
- Metabolic Syndrome or 'Syndrome X' including impaired glucose tolerance, insulin resistance, metabolic acidosis or ketosis, disorders of food intake, satiety disorders, obesity, hyperinsulinemia, dyslipidemia (including hyperlipidemia, hypertriglycerid
- the compounds of the present invention can also be used for the prophylaxis, amelioration and/or treatment of the diseases, disorders and conditions collectively referenced to as "diabetic complications” which include both acute complications and chronic complications.
- disorders and conditions collectively referenced to as "diabetic complications” which include both acute complications and chronic complications.
- acute complications include hyperglycemia (e.g., ketoacidosis), infections (e.g., skin, soft tissue, biliary system, respiratory system and urinary tract infections), etc.
- chronic complications include microangiopathy (e.g., nephropathy, retinopathy), arteriosclerosis (e.g., atherosclerosis, heart infarction, brian infarction, lower extremity arterial occlusion), neuropathy (e.g, sensory nerves, motor nerves, autonomic nerves), foot gangrene, etc.
- Major diabetic complications include diabetic retinopathy, diabetic nephropathy and diabetic neuropathy.
- I for the manufacture of a medicament for the prophylaxis, amelioration and/or treatment of one or more condition(s)/ disease(s)/ disorder(s) involving SGLT-2 inhibition in a subject in need thereof.
- Another embodiment of the present invention provides methods for the prophylaxis, amelioration and/or treatment of one or more condition(s)/ disease(s)/ disorder(s) involving SGLT-2 inhibition in a subject in need thereof that comprises administering a therapeutically effective amount of compound of Formula I.
- compositions comprising the novel compounds of Formula I for the treatment of one or more condition(s)/ disease(s)/ disorder(s) involving SGLT-2 inhibition which comprises administrating to a subject in need thereof a pharmaceutically effective amount of the composition.
- An embodiment of the present invention relates to methods of using the compounds of Formula I of the present invention or compositions comprising the compounds of Formula I for the prophylaxis, amelioration and/or treatment of any one or more condition(s)/ disease(s)/ disorder(s), which comprises administering to a subject in need thereof the compounds of Formula I or compositions comprising a pharmaceutically effective amount of the compounds of Formula I.
- the compounds or their pharmaceutically acceptable salts according to the present invention are useful in the treatment of the aforementioned diseases, disorders and conditions in combination with at least one other therapeutic agent.
- the compounds of the present invention may be used in combination with one or more other therapeutic agents in the treatment, prevention, suppression or amelioration of diseases or conditions for which compounds of the present invention or other therapeutic agents may have utility, where the combination of drugs together are safer or more effective than either drug alone.
- therapeutic agents suitable for combination with the compounds of the present invention include, but are not limited to, known therapeutic agents useful in the treatment of the aforementioned disorders including: anti-diabetic agents; agents for prevention of complications of diabetes; anti-hyperglycemic agents; hypolipidemic/ lipid lowering agents; anti-obesity agents; anti-hypertensive agents, anti-platelet agents, anti- atherosclerotic agents, an ti -inflammatory agents, uricosuric agents, anti-TNF agent or c- AMP raising agents and appetite suppressants.
- the use of the compounds of the present invention in combination with atleast one or more of the aforementioned other therapeutic agents may provide results greater than that possible from each of these medicaments alone or greater than the combined additive effects produced by these medicaments.
- the present compounds and the other therapeutic agents may be administered in the same dosage form or in a separate dosage form by same or different administration route, in dosages and regimens as generally known in the art. Those agents which potentiate the therapeutic effect of SGLT- 2 inhibitors according to the invention may allow the dosage to be reduced.
- Suitable anti-diabetic agents for use in combination with the compounds of the present invention include but are not limited to (a) other SGLT-2 inhibitors; (b) insulin sensitizers including (i) PPAR ⁇ agonists such as thiozolidinediones or glitazones (e.g.
- PPAR ⁇ agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), PPARpan agonists, PPAR ⁇ / ⁇ agonists, PPAR ⁇ / ⁇ dual agonists, PPAR ⁇ / ⁇ dual agonists, PPAR ⁇ antagonists, PPAR ⁇ / ⁇ modulators and PPAR ⁇ / ⁇ / ⁇ modulators, (ii) biguanides such as metformin and phenformin, and (iii) protein tyrosine phosphatase- IB (PTP-IB) inhibitors; (c) insulin or insulin mimetics; (d) sulfonylureas and other insulin secretagogues, such as tolbutamide, chlorpropamide, tolazamide, glyburide (glibenclam), sulfonylureas and other insulin secretagogues, such as tolbutamide,
- suitable agents to be used in combination with the compounds of the present invention, for treatment or prevention of complications of diabetes include but are not limited to GABA-receptor antagonists, Na-channel blockers (e.g. mexiletine hydrochloride, oxacarbazepine or the like), ⁇ -aminobutyric acid receptor antagonists (e.g. topiramat or the like), protein-kinase C inhibitors (e.g. midostaurin or the like), advanced glycation end product inhibitors (e.g. pyridoxamine or the like), transcript factor NF-KB inhibitors (e.g. dexlipotam or the like), lipid peroxide inhibitors (e.g.
- tirilazad mesylate or the like a-linked-acid-dipeptidase inhibitors, carnitine derivatives (e.g. levacecamine, levocarnitine or the like), insulin like growth factor-I, platelet-derived growth factor, platelet-derived growth factor analogues, epidermal growth factor, nerve growth factor, biclomol, sulodexide or aldose reductase inhibitors (e.g. ascorbyl gamolenate, tolrestat, epalrestat or the like).
- carnitine derivatives e.g. levacecamine, levocarnitine or the like
- insulin like growth factor-I e.g. levacecamine, levocarnitine or the like
- platelet-derived growth factor platelet-derived growth factor analogues
- epidermal growth factor e.g. ascorbyl gamolenate, tolrestat, epalrestat or the like
- aldose reductase inhibitors e
- Suitable hypolipidemic/ lipid lowering agents include but are not limited to (a) cholesterol lowering agents such as (i) HMG-CoA reductase inhibitors (lovastatin, simvastatin, pravastatin, cerivastatin, fluvastatin, atorvastatin, itavastatin, and rosuvastatin, and other statins), (ii) bile acid sequestrants (cholestyramine, colestipol, and dialkylaminoalkyl derivatives of a cross-linked dextran), (iii) nicotinyl alcohol, nicotinic acid or a salt thereof, (iv) PPAR agonists as described herein, (v) inhibitors of cholesterol absorption, such as beta-sitosterol and ezetimibe, (vi) acyl CoA cholesterol acyltransferase inhibitors, such as avasimibe, and (vii) anti-oxide inhibitors (i) lovastatin
- fibrates e.g. bezafibrate, fenofibrate, gemfibrozil, clofibrate, ciprofibrate, clinofibrate or the like); (g) MTP inhibitors; (h) squalene synthetase inhibitors and squalene epoxidase inhibitors; (i) upregulators of LDL receptor activity; (j) serum cholesterol lowering agents; (k) thyroid hormone receptor agonists (sodium liothyronine, sodium levothyroxine or the like); (1) carnitine palmitoyltransferase inhibitors (etomoxir or the like); (m) probcol and microsomal triglyceride transfer protein inhibitors.
- MTP inhibitors e.g. bezafibrate, fenofibrate, gemfibrozil, clofibrate, ciprofibrate, clinofibrate or the like
- Suitable anti-obesity compounds include but are not limited to (a) fenfluramine, dexfenfluramine, phenteimine, tetrahydrolipostatin, and the like; (b) neuropeptide Yi or Y 5 antagonists; (c) CB-1 receptor inverse agonists and antagonists; (d) ⁇ 3 adrenergic receptor agonists; (e) melanocortin receptor agonists, in particular melanocortin-4 receptor agonists; (f) ghrelin antagonists; (g) melanin-concentrating hormone (MCH) receptor antagonists; (h) lipase inhibitors like orlistat; (i) serotonin (and dopamine) reuptake inhibitors like sibutramine, topiramate or axokine; (j) thyroid hormone receptor beta drugs; (k) anorectic agents like dexamphetamine, phentermine or mazindol;
- Suitable appetite suppressants for use in combination with the compounds of the present invention include but are not limited to (a) monoamine reuptake inhibitors; (b) dopamine agonists; (c) leptin analogues; (d) ot-melanocyte stimulating hormone; (e) enterostatin agonists; (f) CCK-A agonists; (g) corticotropin releasing hormone; (h) somatostatin; (i) brain-derived neurotrophic factor; (j) orexin receptor agonists.
- Suitable anti-hypertensive agents include but are not limited to (a) vasopeptidase inhibitors like Neutral endopeptidase (neprilysin) inhibitors and/or ACE (angiotensin- converting enzyme) inhibitors or dual NEP/ACE inhibitors (enalapril, lisinopril, captopril, quinapril, trandolapril, fosinpril, benazepril, ramipril, enalaprilat, moexipril or perindopril and the like) and/or PKC inhibitors; (b) beta blockers (like metoprolol, propranolol, atenolol, carvedilol or sotalol) and calcium channel blockers (like amlodipine, diltiazem, nifedipine, verapamil or nicardipine ); (c) Angiopeptidase inhibitors like Neutral endo
- Suitable anti-inflammatory agents for use in combination with the compounds of the present invention include but are not limited to aspirin, non-steroidal anti-inflammatory drugs, glucocorticoids, azulfidine, and selective cyclooxygenase-2 inhibitors.
- Suitable agents to be used in combination with the compounds of the present invention, for the treatment or prevention of hyperuricemia or gout include but are not limited to (a) uric acid synthesis inhibitors e.g. allopurinol, oxypurinol or the like; (b) uricosuric agents e.g. benzbromarone, probenecid or the like; (c) urinary alkaiiizers e.g. sodium hydrogen carbonate, potassium citrate or the like.
- Step 1 Preparation of ((3fl5',5 1 S',6 ⁇ ,6a 1 S -6-((/ert-butyldimethylsilyl)oxy)-2,2-dimethyl tetrahydrofuro[2,3- ⁇ ][l,3]dioxol-5-yl)(3-(4-((tert-butyldirnethylsilyl)oxy)benzyl)-4- chlorophenyl)methanol
- reaction mixture was quenched by the addition of saturated NH 4 C1 solution and extracted with ethyl acetate (3 x 300 mL). The combined organic layers were dried over Na 2 S0 4 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :49 acetone: Pet. Ether) to afford the title compound (3.66 g, 25%)
- Step 2 Preparation of (35,4i?,55,65)-6-(3-(4-acetoxybenzyl)-4-chlorophenyl)tetrahydro- 2H-pyran-2,3,4,5-tetrayl tetraacetate
- Step 3 Preparation of (25',35',4 J R,55,65)-2-bromo-6-(4-chloro-3-(4-hydroxybenzyl) phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- Step 4 Preparation of (25,3 1 S,4 J R,55,65 -2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- methoxytetrahydro-2H-pyran-3 ,4, 5 -triyl triacetate
- Step 5 Preparation of (2S, S, 4R,5S,6S)-2- (3-(4-(2-((ter/-butoxycarbonyl) amino) ethoxy) benzyl)-4-chlorophenyl)-6-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate
- (2S, S, 4R,5S,6S)-2- (3-(4-(2-((ter/-butoxycarbonyl) amino) ethoxy) benzyl)-4-chlorophenyl)-6-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate To a solution of (25,35,4i?,55,65)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate (1.1 g, 2.35 mmol) in dry DMF (10 mL),
- Step 6 Preparation of (2-5',35,4 ?,55 , ,6 l S)-2-(3-(4-(2-((fert-butoxycarbonyl)amino) ethoxy)benzyl)-4-chlorophenyl) -6-methoxytetrahydro-2H-pyran-3 ,4,5-triyl triacetate
- Step 7 Preparation of (2 1 S',35,4i?,55,65)-2-(4-chloro-3-(4-(2-(methylsulfonamido) ethoxy)benzyl)phenyl)-6-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate
- Step 8 Preparation of N-(2-(4-(2-chloro-5-((25,3i?,4 ?,55,65)-3,4,5-trihydroxy-6- methoxytetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)methanesulfonamide
- reaction mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 : 15 MeOH: DCM) to afford title compound (60 mg, 59%) as off-white solid.
- Step 2 Preparation of N-(2-(4-(2-chloro-5-((25,37?,4 ?,55,65)-3,4,5-trihydroxy-6- methoxytetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)sulfamide
- Step 1 Preparation of (25,3i?,45,55,6/?)-2-(3-(4-((1 ⁇ 2ri-butyldimethylsilyl)oxy)benzyl)-4- chlorophenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
- Step 2 Preparation of (2S,3/?,45,5R,6R)-6-(acetoxymethyl)-2-(3-(4-((ieri- butyldimethylsilyl) oxy)benzyl)-4-chlorophenyl)-2-methoxytetrahydro-2H-pyran-3,4,5- triyl triacetate
- Step 3 Preparation of (2i?,3 J R,4i?,55,65)-2-(Acetoxymethyl)-6-(4-chloro-3-(4- hydroxybenzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- Step 5 Preparation of (2/?,3/?,47?,55,65)-2-(acetoxymethyl)-6-(3-(4-(2-((tert- butoxycarbonyl) amino)ethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3 ,4,5-triyl triacetate
- Step 6 Preparation of (2 ?,3i?,4/?,55 , ,65)-2-(acetoxymethyl)-6-(3-(4-(2- aminoethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3 ,4,5-triyl triacetate 2,2,2- trifiuoroacetate
- Step 7 Preparation of (2 ⁇ ,37?,4 J /?,5 ⁇ ,6 , )-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2 (sulfamoylamino)ethoxy)benzyl) phenyl)tetrahydro-2H-pyran-3 ,4,5-triyl triacetate
- Step 8 Preparation of (25,3i?,4i?,55,6i?)-2-(3-(4-(2-(sulfamoylamino)ethoxy)benzyl)-4- chlorophenyl)-tetrahydro-6-(hydroxymethyl)-2H-pyran-3 ,4,5-triol
- Step 3 Preparation of (2i3 ⁇ 4,3 ⁇ ,4 ⁇ ,5S,6S)-2-(acetox methyl)-6-(3-(4-(3-((tert- butoxycarbonyl) amino)propoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- Step 4 Preparation of (2 ?,3i?,4 ?,55,65)-2-(acetoxymethyl)-6-(3-(4-(3- aminopropoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2,2,2- trifluoroacetate
- Step 6 Preparation of (25',3/?,4i?,55,6 ?)-2-(3-(4-(3-(sulfamoylamino)propoxy)benzyl)-4- chlorophenyl)-tetrahydro-6-(hydroxymethyl)-2H-pyran-3,4,5-triol
- Step 1 Preparation of (2i?,3i?,4i?,55,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(3- (cyclohexanesulfonamido) propoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- Step 2 Preparation of N-(3-(4-(2-chloro-5-((25,3 ?,4i?,55,6i?)-3,4,5-trihydroxy-6-
- Step 1 Preparation of (2/?,3 ⁇ ,4 ⁇ ,55,65)-2-(3 ⁇ 6 ⁇ 1)-6-(4- ⁇ 1 ⁇ 1 ⁇ -3-(4-(2-)
- reaction mixture was diluted with DCM (50 mL) and given water washings (2 x 10 mL). The organic layer was dried over anhydrous Na 2 S0 4 . Volatiles were evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 2.5:97.5 MeOH: DCM) to afford title compound as a white solid (180 mg 3 ⁇ 4 57.01%).
- Step 2 Preparation of JV-(2-(4-(2-chloro-5-((2,S,3i?,4 J R,55,6 J R)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)- , J P- dimethylphosphinic amide
- reaction mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :10 MeOH: DCM) to afford title compound (92mg, 72.5%) as off-white solid.
- Step 3 Preparation of dimethyl (3-(4-(2-chloro-5-((25,37i,4/?,55,6 ?)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)propyl)phosphoramidate
- reaction mixture was stirred at the same temperature and progress of the reaction was monitored by TLC. On completion, the resulting mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide title compound (100 mg, 35 %) as off white solid.
- Step 4.1 To a solution of l-(2-(Allyloxy)ethyl)-4-bromobenzene (1 g, 4.14 mmol) in dry
- Step 4.2 To a solution of (4-(2-(Allyloxy)ethyl)phenyl)(5-bromo-2- chlorophenyl)methanol (5.1 g, 13.36 mmol) in acetonitrile (40 mL) triethylsilane (6.82 mL, 42.75 mmol) was added at 10°C followed by the addition of boranetrifluoroetherate (3.35 mL, 26.72 mmol). The resulting mixture was stirred at r.t. for 4 h.
- Step 5.1 To a solution of 2-(4-(2-(allyloxy)ethyl)benzyl)-4-bromo-l-chlorobenzene (300 mg, 0.818 mmol) in dry THF (5 mL), "BuLi (0.82 mL, 1.23 mmol, 1.5 M in hexane) was added at -78 °C and allowed to stir for 5 min.
- Step 5.2 To a solution of (25,3i?,45,55,6 ?)-2-(3-(4-(2-(allyloxy)ethyl)benzyl)-4- chlorophenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (3.86 g, 8.05 mmol) in dry THF (42 mL), diisopropylethylamine (10.29 mL, 59.07 mmol), 4- •dimethylaminopyridine (354 mg, 2.89 mmol) were added. Acetic anhydride (4.94 mL, 52.32 mmol) was added to the resulting solution at 0 °C.
- Step 6 Preparation of (2i?,3i?,4i?,5S,6S)-2-(acetoxymethyl)-6-(3-(4-(2- (allyloxy)ethyl)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- Step 7 Preparation of (2i?,3i?,4/?,5 1 S,65 -2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- hydroxyethyl)benzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate
- Step 8 Preparation of (2 ?,3R,4R,5S,6S)-2-(acetox methyl)-6-(4-chloro-3-(4-(2- ⁇ ((methylsulfonyl)oxy)ethyl)benzyl)phenyl)tetrahydro-2H-pyran-3 ,4,5 -triyl triacetate
- reaction mixture was diluted with DCM (20 mL), washed with water (10 mL) and brine (10 mL) successively. The organic layer was dried over anhydrous Na 2 S0 4 and the volatiles were evaporated in vacuo to afford the title compound which was used as such in further steps.
- Step 10 Preparation of (2i?,3i?,4 ,5S,6S)-2-(acetoxymethyl)-6-(3-(4-(2- aminoethyl)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- Step 11 Preparation of (2 ⁇ ,3 ⁇ ,4 ⁇ ,5- , ,65 , )-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- (sulfamoylamino)ethyl)benzyl)phenyl) tetrahydro-2H-pyran-3 ,4,5-triyl triacetate
- Step 1 Preparation of (2 ?,3/?,4 ⁇ ,55,65)-2-(acetoxymethyl)-6-(3-(4-(2- aminoethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- reaction mixture was diluted with ethyl acetate (20 mL), washed with 5 % HC1 (2 x 10 mL), water (10 mL) and brine (10 mL) successively.
- the organic layer was dried over anhydrous Na 2 S0 4 and the volatiles were evaporated in vacuo.
- the residue obtained was purified by column chromatography (silica gel, 3:7 ethylacetate: Pet. ether) to provide the title compound (80 mg, 65.5 %) as white solid.
- Step 3 Preparation of N-(2-(4-(2-chloro-5-((25,3i?,4i?,55,6i?)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)pyrrolidine-l- sulfonamide
- Step 1 Preparation of ((2i?,35,4i?,5 J R,65)-6-(4-chloro-3-(4-(2-(sulfamoylamino)ethoxy) benzyl)phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methyl 4- methylbenzenesulfonate
- reaction mixture was diluted with ethyl acetate (50 mL), washed with 5% HCl (2 x 20 mL), water (20 mL) and brine (20 mL) successively.
- the organic layer was dried over anhydrous Na 2 S0 and the volatiles were evaporated in vacuo.
- the residue obtained was purified by column chromatography (silica gel, 1 :4 MeOH: DCM) to provide the title compound (605 mg, 73 %) as white solid.
- Step 2 Preparation of ((2 J R,35,4i?,5i?,65)-6-(4-chloro-3-(4-(2-(sulfamoylamino) ethoxy)benzyl)phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methylazide
- Step 3 Preparation of ((2/?,35',4i?,5i?,65)-6-(4-chloro-3-(4-(2-(sulfamoylamino)ethoxy) benzyl)phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methylamine
- ((2i?,35,4i?,5 ?,6 l S)-6-(4-cliloro-3-(4-(2-(sulfamoylamino)ethoxy) benzyl) phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methylazide 440 mg, 0.833 mmol
- triphenylphosphine 328 mg, 1.24 mmol
- Step 4 Preparation of (25,3i?,4i?,55,6i?)-2-(3-(4-(2-(sulfamoylamino)ethoxy)benzyl)-4- chlorophenyl)-tetrahydro-6-((3-cyclobutyl ureido) methyl)-2H-pyran-3,4,5-triol
- Step 4.1 To a solution of cyclobutylamine (200 mg, 2.81 mmol) in dry DCM (14 mL), carbonyldiimidazole (755 mg, 4.21 mmol) was added at 0 °C. The resulting mixture was stirred at r.t. for 2h. Reaction mixture was diluted with DCM (25 mL) and washed with water (20 mL) and brine (20 mL) successively. Organic layer was dried over anhydrous Na 2 S0 4 and the volatiles were evaporated in vacuo to obtain N-cyclobutyl-lH-imidazoIe- 1 -carboxamide as crude mixture. This was used as such in the next step.
- Step 4.2 To a solution of ((2i?,35',4 J R,5/?,6 1 S)-6-(4-chloro-3-(4-(2-(sulfamoylamino)ethoxy) benzyl )phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methylamine (120 mg, 0.24 mmol) in dry THF (1.15 mL), N-cyclobutyl-lH-imidazole-1 -carboxamide (39.4 mg, 0.24 mmol) was added at r.t. The resulting mixture was stirred at r.t. for 16 h and the reaction was monitored by TLC.
- reaction mixture was diluted with ethyl acetate (20 mL), washed with water (10 mL) and brine (10 mL) successively.
- the organic layer was dried over anhydrous Na 2 S0 4 and the volatiles were evaporated in vacuo.
- the residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide the title compound (130 mg, 91 %) as white solid.
- Step 1 Preparation of (2R,3R,4R,5S,6S)-2-(acetoxymethy ⁇ )-6-(3-(4-((tert- butyldimethylsilyl)oxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- (2R,3R,4R,5S,6S)-2-(acetoxymethy ⁇ )-6-(3-(4-((tert- butyldimethylsilyl)oxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 500 mg, 0.91 mmol) in dry DCM (2.70 mL), triethyl amine (0.32 mL, 2.27 mmol) and 4-dimethylaminopyridine (10 mg, 0.08 mmol) were added at 0 °C followed by the addition of tert- buty
- reaction mixture was stirred at r.t. for 16 h and the reaction was monitored by TLC. On completion, reaction mixture was diluted with DCM (20 mL), washed with water (10 mL) and brine (10 mL) successively. The organic layer was dried over anhydrous Na 2 S0 4 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 acetone: Pet. ether) to provide the title compound (530 mg, 87.74 %) as white solid.
- Step 2 Preparation of (2S,3/?,4i?,55,6i?)-2-(3-(4-((iert-butyldimethylsilyl)oxy)benzyl)-4- chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
- Step 3 Preparation of ((27?,3 1 S',4i?,5i?,6 i S -6-(3-(4-((fer/-butyldimethylsilyl)oxy)benzyl)-4- chlorophenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methy]-4- methylbenzenesulfonate
- reaction mixture was diluted with ethyl acetate (50 mL), washed with 5% HC1 (2 x 20 mL), water (20 mL) and brine (20 mL) successively.
- the organic layer was dried over anhydrous Na 2 S0 4 and the volatiles were evaporated in vacuo.
- the residue obtained was purified by column chromatography (silica gel, 1 :9 acetone: DCM) to provide the title compound (400 mg, 89.9 %) as white solid.
- Step 4 Preparation of (25 , ,3i?,4/?,55',6i?)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- ((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-3,4,5-triol
- Step 5 Preparation of tert-butyl (4-(4-(2-chloro-5-((2 1 S,3 ?,4 J R,55,6i?)-3,4,5-trihydroxy-6- ((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)butyl)caribamate
- tert-butyl 4-(4-(2-chloro-5-((2 1 S,3 ?,4 J R,55,6i?)-3,4,5-trihydroxy-6- ((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)butyl)caribamate
- reaction mixture was stirred at r.t. for 1 h and the reaction was monitored by TLC. On completion, reaction mixture was diluted with ethylacetate (20 mL), washed with water (10 mL) and brine (10 mL) successively. The organic layer was dried over anhydrous Na 2 S0 4 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 3:7 acetone: Pet. ether) to provide the title compound (410 mg, 95.12 %) as white solid.
- Step 7 Preparation of (25,35,4i?,5i?,6/?)-2-(3-(4-(4-aminobutoxy)benzyl)-4- chlorophenyl)-6-((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2,2,2-trifluoroacetate
- Step 8 Preparation of (25,35,4ii,5 J R,6i?)-2-(4-chloro-3-(4-(4-(sulfamoylamino)butoxy) benzyl)phenyl)-6-((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- 2,2,2-trifluoroacetate 250 mg, 0.323 mmol
- triethylamine (0.14 mL, 0.97 mmol
- Step 9 Preparation of (2/?,3 l S',4v , ?,57?,65)-2-((2,2,2-trifluoroethoxy)methyl)-6-(3-(4-(4- aminosulfonylaminobutoxy)benzyl)-4-chlorophenyl)-tetrahydro-2H-pyran-3,4,5-triol
- Step 1 Preparation of (2i?,37?,4i?,55,65)-2-(acetoxymethyl)-6-(3-(4-(2-((N- acetylsulfamoyl) amino)ethoxy) benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- reaction mixture was diluted with EtOAc (150 mL) and washed with water (5 x 10 mL). The organic layer was dried over anhydrous Na 2 S0 4 and volatiles were removed in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :24 MeOH: DCM) to afford title compound (100 mg, 55.0%) as a white solid.
- Step 2 Preparation of N-(N-(2-(4-(2-chloro-5-((25,3 ?,4i?,55,6i?)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)sulfamoyl)acetamide
- (2/?,3R,4R,55,65)-2-(acetoxymethyl)-6-(3-(4-(2-(( ⁇ - acetylsulfamoyl) amino)ethoxy) benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 150 mg, 0.21 mmol
- MeOH:THF:Water (3:2: 1, 2.1 mL) was added lithium hydroxide monohydrate (8.8 mg, 0.210 mmol) at 0 °C .
- reaction mixture was stirred at r.t. for 1 h. After completion of reaction, as confirmed by TLC, the reaction mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to afford the title compound (95mg, 83%) as white solid.
- Step 1 Preparation of (2 J ,3i?,4 J R,5S,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2-((N,N- dimethylsulfamoyl)(methyl)amino)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- (2i?,3i?,4i?,5S,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2 (sulfamoylamino)ethoxy)benzyl) phenyl )tetrahydro-2H-pyran-3,4,5-triyl triacetate 1 10 mg, 0.16 mmol) in dry DMF (3 mL), cesium carbonate (213 mg, 0.67 mmol) was added followed by the addition of methyl iodide (0.03 mL, 0.49 mmol) at 0
- reaction was stirred at r.t. for 24 h. After completion, as confirmed by TLC, the reaction mixture was diluted with ethylacetate (150 mL) and washed with water (2 x 20 mL). The organic layer was dried over anhydrous Na 2 S0 4 volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 4:6 acetone: Pet. ether) to afford title compound as a white solid (105mg, 98.32%).
- Step 2 Preparation of N-(2-(4-(2-chloro-5-((2S,3i?,4i?,55,6/?)-3,4,5-trihydroxy-6-
- Step 1 Preparation of (2i?,3i?,4i?,5S,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- (methylsulfonamido)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- Step 2 Preparation of N-(2-(4-(2-chloro-5-((25,3 ⁇ ,4 ⁇ ,5 ⁇ ,6 ⁇ )-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)methanesulfonamide
- the title compound (350 mg, 9.45 %) was synthesized following the procedure reported for the synthesis of (25 , ,3 ⁇ ,4 ?,5.S , ,6 ⁇ )-2-(3-(4-(2- (sulfamoylamino)ethoxy)benzyl)-4-chlorophenyl)-tetrahydro-6-(hydroxymethyl)-2H- pyran-3,4,5-triol starting with (2i?,3i?,4 ?,55,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- (methylsulfonamido)ethoxy)benzyl)pheny
- Step 3 Preparation of ((2i ! ?,3 1 S',4 ⁇ ,5 ⁇ ,6 l )-6-(4-chloro-3-(4-(2-(methylsulfonamido) ethoxy)benzyl)phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methyl-4- methylbenzenesulfonate
- Step 4 Preparation of N-(2-(4-(2-chloro-5-((25,3 ?,4i?,55,6i?)-3,4,5- trihydroxy-6- (piperidin- 1 -ylmethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)
- reaction mixture was diluted with ethylacetate (150 mL) and washed with water (3 x 10 mL). The organic layer was dried over anhydrous Na 2 S0 4 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 Acetone: Pet. ether) to afford title compound (392 mg, 60.85%) as white solid.
- Step 3 Preparation of (2 ?,3 ⁇ ,4i?,5 ⁇ ,65)-2-(acetoxymethyI)-6-(4-chloro-3-(4-(2- hydroxyethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- reaction mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 2:3 EtOAc: Pet ether) to afford title compound (1.8g, 82 %) as a liquid.
- Step 4 Preparation of (2/?,3i?,4 ?,55',65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- (tosyloxy)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- reaction mixture was diluted with ethyl acetate (150 mL) and washed with 2N HC1 (3 x 20 mL). The organic layer was dried over anhydrous Na 2 S0 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 : 1 EtOAc: Pet. ether) to afford title compound (130 mg, 35%) as white solid.
- Step 5 Preparation of (2i?,3i?,4 ⁇ ,55,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2-(4- oxido-4-phenyl-l ,4-azaphosphinan-l-yl)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5- triyl triacetate
- reaction mixture was stirred at 80 °C for 36 h. After completion of reaction, as confirmed by TLC, the reaction mixture was diluted with EtOAc (150 mL) and washed with water (3 x 20 mL). The organic layer was dried over anhydrous Na 2 S0 4 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:48 MeOH: DCM) to afford title compound (65mg, 63.1%) as a white solid.
- Step 6 Preparation of l-(2-(4-(2-chloro-5-((25,3 ⁇ ,4 ?,55,6i?)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)-4-phenyl-l ,4- azaphosphinane 4-oxide
- reaction mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 3:7 MeOH: DCM) to afford title compound (58 mg, 80.3%) as off white solid.
- Mouse SGLT-2 cDNA was amplified from C57BL/6J mouse kidneys and introduced in the pcDNA3.1(+) expression vector (Invitrogen, Inc.) and propagated in Escherichia coli strain DH5a using Luria-Bertani (LB) medium containing ampicillin.
- Mouse SGLT-2 recombinant expression plasmid DNA was transfected into CHO-K1/HEK cells (American Type Culture Collection) using Superfect Transfection Reagent according to a manufacturer suggested protocol. Stably transfected cells were selected using G418 antibiotic selection pressure.
- Cells expressing mSGLT-2 were seeded on 96-well tissue culture plates (Greiner, Inc.) in RPMI containing 10% FBS and 400 ⁇ g/mL G418 (0.8 x 10 5 cells per well in 200 ⁇ medium) and incubated at 37 °C under 5% carbon dioxide for 24 h prior to the assay.
- HEK cells HEK cells
- HEK cells HEK cells
- HEK cells HEK cells
- HEK cells HEK cells
- HEK cells HEK cells
- HEK cells HEK cells
- HEK cells HEK cells
- HEK cells HEK cells
- HEK cells HEK cells
- HEK cells HEK cells
- HEK cells HEK cells
- HEK cells HEK cells
- Human SGLT-1 cDNA in the pCMV-XL-Neo expression vector was obtained from Origene Corporation and propagated in Escherichia coli strain DH5a using Luria- Bertani (LB) medium containing ampicillin.
- Human SGLT-1 expression plasmid DNA was transfected into CHO-Kl cells (American Type Culture Collection) using Superfect Transfection Reagent according to a manufacturer suggested protocol. Stably transfected cells were selected using G418 antibiotic selection pressure.
Abstract
The present invention relates to novel compounds of Formula (I), their pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof. The invention also relates to the processes for the synthesis of novel compounds of Formula I, their pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof. The present invention also provides pharmaceutical compositions comprising novel compounds of Formula I and methods of treating or preventing one or more conditions or diseases that may be regulated or normalized via inhibition of Sodium Glucose Cotransporter-2 (SGLT-2). The invention also relates to the use of compounds of Formula I, their pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, for the manufacture of a medicament for the prophylaxis, amelioration and/or treatment of conditions or diseases that may be regulated or normalized via inhibition of Sodium Glucose Cotransporter-2 (SGLT-2) and the related diseases, disorders and conditions, in a subject in need thereof.
Description
NOVEL SUGAR DERIVATIVES
Field of the invention
The present invention relates to novel compounds of Formula I, their pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof. The invention also relates to the processes for the synthesis of novel compounds of Formula I, their pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof. The present invention also provides pharmaceutical compositions comprising novel compounds of Formula I and methods of treating or preventing one or more conditions or diseases that may be regulated or normalized via inhibition of Sodium Glucose Cotransporter-2 (SGLT-2). The invention also relates to the use of compounds of Formula I, their pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, for the manufacture of a medicament for the prophylaxis, amelioration and/or treatment of conditions or diseases that may be regulated or normalized via inhibition of Sodium Glucose Cotransporter-2 (SGLT-2) and the related diseases, disorders and conditions, in a subject in need thereof. Background of the invention
Diabetes is a metabolic disorder which is rapidly emerging as a global health care problem that threatens to reach pandemic levels. The number of people with diabetes worldwide is expected to rise from 285 million in 2009 to 435 million by 2030. Diabetes results from deficiency in insulin because of impaired pancreatic β-cell function or from resistance to insulin in body, thus leading to abnormally high levels of blood glucose.
Diabetes which results from complete deficiency in insulin secretion is type 1 diabetes and the diabetes due to resistance to insulin activity together with an inadequate insulin secretion is type 2 diabetes. Type 2 diabetes (Non insulin dependent diabetes) accounts for 90-95 % of all diabetes. An early defect in type 2 diabetes mellitus is insulin resistance which is a state of reduced responsiveness to circulating concentrations of insulin and is often present years before clinical diagnosis of diabetes. A key component of the pathophysiology of type 2 diabetes mellitus involves an impaired pancreatic β-cell function which eventually contributes to decreased insulin secretion in response to elevated plasma glucose. The β-cell compensates for insulin resistance by increasing the
insulin secretion, eventually resulting in reduced β-cell mass. Consequently, blood glucose levels stay at abnormally high levels (hyperglycemia).
Hyperglycemia is central to both the vascular consequences of diabetes and the progressive nature of the disease itself. Chronic hyperglycemia leads to decrease in insulin secretion and further to decrease in insulin sensitivity. As a result, the blood glucose concentration is increased, leading to diabetes, which is self-exacerbated. Chronic hyperglycemia has been shown to result in higher protein glycation, cell apoptosis and increased oxidative stress; leading to complications such as cardiovascular disease, stroke, nephropathy, retinopathy (leading to visual impairment or blindness), neuropathy, hypertension, dyslipidemia, premature atherosclerosis, diabetic foot ulcer and obesity. So, when a person suffers from diabetes, it becomes important to control the blood glucose level. Normalization of plasma glucose in type 2 diabetes patients improves insulin action and may offset the development of beta cell failure and diabetic complications in the advanced stages of the disease.
Diabetes is basically treated by diet and exercise therapies. However, when sufficient relief is not obtained by these therapies, medicament is prescribed alongwith. Various anti-diabetic agents being currently used include biguanides (decrease glucose production in the liver and increase sensitivity to insulin), sulfonylureas and meglitinides (stimulate insulin production), a-glucosidase inhibitors (slow down starch absorption and glucose production) and thiazolidinediones (increase insulin sensitivity). These therapies have various side effects: biguanides cause lactic acidosis, sulfonylurea compounds cause significant hypoglycemia, a-glucosidase inhibitors cause abdominal bloating and diarrhea, and thiazolidinediones cause edema and weight gain. Recently introduced line of therapy includes inhibitors of dipeptidyl peptidase-IV (DPP-IV) enzyme, which may be useful in the treatment of diabetes, particularly in type 2 diabetes. DPP-IV inhibitors lead to decrease in inactivation of incretins glucagon like peptide- 1 (GLP-1) and gastric inhibitory peptide (GIP), thus leading to increased production of insulin by the pancreas in a glucose dependent manner. All of these therapies discussed, have an insulin dependent mechanism.
Another mechanism which offers insulin independent means of reducing glycemic levels is the inhibition of sodium glucose co-transporters (SGLTs). In healthy individuals, almost 99% of the plasma glucose filtered in the kidneys is reabsorbed, thus leading to only less than 1% of the total filtered glucose being excreted in urine. Two types of SGLTs, SGLT-1 and SGLT-2, enable the kidneys to recover filtered glucose. SGLT-1 is a
low capacity, high-affinity transporter expressed in the gut (small intestine epithelium), heart, and kidney (S3 segment of the renal proximal tubule), whereas SGLT-2 (a 672 amino acid protein containing 14 membrane-spanning segments), is a low affinity, high capacity glucose transporter, located mainly in the SI segment of the proximal tubule of the kidney. SGLT-2 facilitates approximately 90% of glucose reabsorption and the rate of glucose filtration increases proportionally as the glycemic level increases. The inhibition of SGLT-2 should be highly selective, because non-selective inhibition leads to complications such as severe, sometimes fatal diarrhea, dehydration, peripheral insulin resistance, hypoglycemia in CNS and an impaired glucose uptake in the intestine.
Humans lacking a functional SGLT-2 gene appear to live normal lives, other than exhibiting copious glucose excretion with no adverse effects on carbohydrate metabolism. However, humans with SGLT-1 gene mutations are unable to transport glucose or galactose normally across the intestinal wall, resulting in condition known as glucose- galactose malabsorption syndrome.
Hence, competitive inhibition of SGLT-2, leading to renal excretion of glucose represents an attractive approach to normalize the high blood glucose associated with diabetes. Lower blood glucose levels would, in turn, lead to reduced rates of protein glycation, improved insulin sensitivity in liver and peripheral tissues, and improved cell function. As a consequence of progressive reduction in hepatic insulin resistance and the elevated hepatic glucose output, which are characteristic of type 2 diabetes, would also be expected to gradually diminish to normal values. In addition, excretion of glucose may reduce overall caloric load and lead to weight loss. Risk of hypoglycemia associated with SGLT-2 inhibition mechanism is low, because there is no interference with the normal counter regulatory mechanisms for glucose.
The first known non-selective SGLT-2 inhibitor was the natural product phlorizin
(l-[2,4-dihydroxy-6-[(25 R,4R,55,6R)-3,4,5-trihydrpxy-6-
(hydroxymethyl)tetrahydropyran-2-yl]oxy-phenyl]-3-(4-hydroxyphenyl)propan-l-one). Subsequently, several other synthetic analogues were derived based on the structure of phlorizin. Optimisation of the scaffolds to achieve selective SGLT-2 inhibitors led to the discovery of several considerably different scaffolds, which include C-glycoside derivatives and O-glycoside derivatives.
C-glycoside derivatives have been disclosed, for example, in PCT publications WO2004013118, WO2005085265, WO2006008038, WO2006034489, WO2006037537, WO2006010557, WO2006089872, WO2006002912, WO2006054629, WO2006064033,
WO20071361 16, WO2007000445, WO2007093610, WO2008069327, WO200802001 1, WO2008013321, WO2008013277, WO2008122014, WO2008116195, WO2008042688, WO2009026537 and WO2010022313, US patents US6515117B2, US6936590B2 and US7202350B2 and Japanese patent application JP2004359630. O-glycoside derivatives have been disclosed, for example, in PCT publications WO2002088157, WO2002064606, WO2003020737, WO2003000712, WO2004089966, WO2004058790, WO2004099230, WO2004087727, WO2005085267, WO2005095429, WO2005021566, WO200601 1469 and WO20071261 17 and US patents US6555519B2, US6683056B2, US6872706B2, US7056892B2, US7129381B2, US7189702B2, US7247616B2 and US7294618B2.
The compounds shown below are the SGLT-2 inhibitors which have reached advanced stages of human clinical trials: Bristol-Myers Squibb's "Dapagliflozin" with Formula A and Mitsubishi Tanabe and Johnson & Johnson's "Canagliflozin" with Formula B. Various other compounds whose structures are not disclosed yet but are known to be in different phases of clinical trials are: Lexicon's Lx 421 1, Boehringer's BI 10773,

Formula A Formula B
In spite of all these molecules in advanced stages of human clinical trials, there is still no drug available in the market as SGLT-2 inhibitor. Out of the potential candidates entering the clinical stages, many have been discontinued, emphasizing the unmet need. Thus, there is an ongoing requirement to screen more scaffolds useful as SGLT-2 inhibitors that can have advantageous potency, stability, selectivity, better half-life, and/ or better pharmacodynamic properties. In this regard, novel SGLT-2 inhibitors are provided herein.
Summary of the invention
The present invention relates to the novel compounds of Formula I,
- A -

Formula I
their pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, wherein:
ring A represents aryl;
ring B represents either aryl or heteroaryl;
U, V and W are independently selected from -OH, hydrogen, halogen, C] -i2alkoxy, -CN, -(CH2)„NR8R9, -OR8, -C(=Y)OR8 or -C(=Y)NR8R9; provided that atleast two groups out of U, V and W represent -OR8;
Y represents either O or S;
R7 is selected from halogen, Ci-i2alkyl, C2-i2alkenyl, C2-i2alkynyl, d-nalkylcarbonyl, CM2 alkoxycarbonyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)nRe, -CN, -N02, - NR8R9, -N3, -CR8(=NOR9), -OH, -OR8, -CH2OH, -C(=Y)R8, -C(=Y)OR8, -C(=Y)SR8, - C(=Y)NR8R9, -OC(=Y)R8, -OC(=Y)OR8, -OC(=Y)NR8R9, -OP(=0)R8R9, -(CH2)n- heterocyclyl, -(CH2)n-NR8R9, -(CH2)n-N3, -(CH2)n-NCS, -(CH2)n-S(0)dR8, -(CH2)n- S(0)dNR8R9, -(CH2)n-P(=0)R8R9, -(CH2)n -0P(=O)R8R9, -(CH2)n-NR8C(=Y)R9, -(C¾)n- NR8C(=Y)OR9, -(CH2)n-NR10C(=Y)NR8R9,
-(CH2)„ -NR8S(0)dR9 or -(CH2)n-NHP(=0)R8R9 ; each of which may optionally be substituted at any available position by one or more substituents selected from R1 ';
R1, R2, R3 and R4 are independently selected from hydrogen, halogen, Ci_i2alkyl, C2.]2 alkenyl, C2-i2alkynyl,
 C2_i2haloalkenyl, C2_i2haloalkynyl, Ci-i2alkoxy, Ci_
C2_i2haloalkenyl, C2_i2haloalkynyl, Ci-i2alkoxy, Ci_
 C]_i2alkylcarbonyl, Ci-12 alkoxycarbonyl, C -2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, cycloalkenyl, cycloalkynyl, -(CH2)„-heterocyclyl, -(CH2)n-aryl, -(CH2)n-heteroary], - CN, -N02, -NR12R13, -(CH2)nNRl2R13, -N3, -NCO, -(CH2)nN3, -(CH2)„ NCS, - CR12(=NOR13), -NR14NR12R13, oxo, -OR12, -SR12, -(CH2)nYR12, -S(0)dR12, - S(0)dNR12R13, -(CH2)nS(0)dR12, -(C¾)nS(0)dNRi2R!3, -P(=0)R12R13, - (CH2)nP(=0)R12R13, -C(=Y)R12, -C(=Y)OR12, -C(=Y)SR12, -C(=Y)NR,2R13, -
C]_i2alkylcarbonyl, Ci-12 alkoxycarbonyl, C -2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, cycloalkenyl, cycloalkynyl, -(CH2)„-heterocyclyl, -(CH2)n-aryl, -(CH2)n-heteroary], - CN, -N02, -NR12R13, -(CH2)nNRl2R13, -N3, -NCO, -(CH2)nN3, -(CH2)„ NCS, - CR12(=NOR13), -NR14NR12R13, oxo, -OR12, -SR12, -(CH2)nYR12, -S(0)dR12, - S(0)dNR12R13, -(CH2)nS(0)dR12, -(C¾)nS(0)dNRi2R!3, -P(=0)R12R13, - (CH2)nP(=0)R12R13, -C(=Y)R12, -C(=Y)OR12, -C(=Y)SR12, -C(=Y)NR,2R13, -

OC(=Y)R12, -OC(=Y)OR12, -OC(=Y)NR!2RB,-OP(=0)R,2R13, -(CH2)nOC(=Y)R12 , -
(CH2)nOC(=Y)OR12, -(CH2)nOC(=Y)NR12R13, -(CH2)nOP(=0)R,2R13, -N(R12)C(=Y)R13, -N(Rl2)C(=Y)OR13, -N(R14)C(=Y)NR12R13, -NR, S(0)dR13, -NHP(=0)R12R13, - (CH2)nNRI2C(=Y)R13, -(CH2)nNR12C(=Y)OR13, -(CH2)nNR14C(=Y)NR12R13, - (CH2)nNR,2S(0)dR13 or -(CH2)nNHP(=0)R12R13; each of which may optionally be substituted at any available position by one or more substituents selected from R1 1 ;
L is selected from O, S, SO, S02, -C(=0)-, -(CH2)„-, -C(=CH2)-, 1 ,1-cyclopropylene, -
1 R
NR - or -(C(R )2)m- ; each methylene group may optionally be substituted with one or more substituents independently selected from halogen, hydroxy, oxo, -C(=0)0-, - C(=0)NR16-, Ci-12alkyl, C1-12alkoxy, -C3-20cycloalkyl or -C3-2ocycloalkoxy ;
E can be absent or is selected from CH2, O, S or NRi6;
G can be absent or is selected from Ci-i2alkylene, C2-i2alkenylene, C2-i2alkynylene, C i2 alkylenecarbonyl, C3-20cycloalkylene, heterocyclyl, aryl, heteroaryl, -NR15-, - (CH2)nNR15-, -(CH2)nS(0)d-, -(CH2)nS(0)d NR15-, -(CH2)nP(=0)R15-, -C(=Y)-, C(=Y)NR15-, -(CH2)„C(=Y)-, -(CH2)nC(=Y)NR15-, -(CH2)nOC(=Y)- , (CH2)nOP(=0)R15- or -(CH2)nNR15S(0)d-; each of which may optionally be substituted at any available position by one or more substituents selected from R1 1;
R5 and R6 are independently selected from hydrogen, CM2alkyl, -S(0)dRa, -S(0)dNRaRb or -P(=0)R Rb; each of which may optionally be substituted at any available position by R11; wherein Ra and Rb can be joined together to form a monocyclic or polycyclic ring, which may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R 1 1 ;
or
R5 and R6 are joined together along with the nitrogen atom to which they are attached to form a monocyclic or polycyclic ring, which contains atleast one phosphorus atom and may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R1 1 ;
provided that
(a) both R5 and R6 can not be hydrogen at the same time
(b) both R5 and R6 can not be alkyl at the same time
(c) R5 and R6 can not be a combination of hydrogen and alkyl at the same time
(d) when E and G are absent and R5 is hydrogen then R6 can not represent -S(0)dRa
(e) when R7 represents
 C2-i2alkenyl, C2-i2alkynyl, -CH2OH or -(CH2)nRe, wherein n is not equal to zero; one of R5 and R6 represents -H or Ci-6alkyl and the other represents -S(0)dRa, wherein d represents 1 or 2; then Ra can not be C\. 6alkyl, C2.6alkenyl, C2-6alkynyl, aryl or heteroaryl;
C2-i2alkenyl, C2-i2alkynyl, -CH2OH or -(CH2)nRe, wherein n is not equal to zero; one of R5 and R6 represents -H or Ci-6alkyl and the other represents -S(0)dRa, wherein d represents 1 or 2; then Ra can not be C\. 6alkyl, C2.6alkenyl, C2-6alkynyl, aryl or heteroaryl;
R8, R9, R10, R12, R13, R14 and R15 are independently selected from hydrogen, halogen, Cj . nalkyl, C2-i2alkenyl, C2.i alkynyl,
 Ci_i2alkoxycarbonyl, C3. 20cycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, -(CH2)n-heterocyclyl, - (CH2)n-aryl, -(CH2)n-heteroaryl, -CN, -N02; -NRaRb, -(CH2)nNRaRb, -N3, -NCS, - (CH2)nN3, -(CH2)„ NCS, -CRa(=NORb), -NRcNRaRb, -ORa, -SRa, -(CH2)nYRa, -S(0)dRa, -S(0)d NRaRb, -(CH2)nS(0)dRa, -(CH2)n S(0)d NRaRb, -P(=0)RaRb, -(CH2)n P(=0)RaRb, -C(=Y)Ra, -C(=Y)ORa, -C(=Y)SRa, -C(=Y)NRaRb, -(CH2)nC(=Y)Ra, - (CH2)nC(=Y)ORa, -(CH2)nC(=Y)NRaRb, -(CH2)n-C(=Y)SRa, -OC(=Y)Ra, -OC(=Y)ORa, - OC(=Y)NRaRb, -OP(=0)RaRb, -(CH2)nOC(=Y)Ra , -(CH2)nOC(=Y)ORa, - (CH2)nOC(=Y)NRaRb, -(CH2)nOP(=0)RaRb, -N(Ra)C(=Y)Rb, -N(Ra)C(=Y)ORb, - N(R°)C(=Y)NRaRb, -NRaS(0)d Rb, -NHP(=0)RaRb, -(CH2)nNRaC(=Y)Rb, - (CH2)nNRaC(=Y)ORb, -(CH2)nNRcC(=Y)NRaRb, -(CH2)nNRaS(0)dRb or -
Ci_i2alkoxycarbonyl, C3. 20cycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, -(CH2)n-heterocyclyl, - (CH2)n-aryl, -(CH2)n-heteroaryl, -CN, -N02; -NRaRb, -(CH2)nNRaRb, -N3, -NCS, - (CH2)nN3, -(CH2)„ NCS, -CRa(=NORb), -NRcNRaRb, -ORa, -SRa, -(CH2)nYRa, -S(0)dRa, -S(0)d NRaRb, -(CH2)nS(0)dRa, -(CH2)n S(0)d NRaRb, -P(=0)RaRb, -(CH2)n P(=0)RaRb, -C(=Y)Ra, -C(=Y)ORa, -C(=Y)SRa, -C(=Y)NRaRb, -(CH2)nC(=Y)Ra, - (CH2)nC(=Y)ORa, -(CH2)nC(=Y)NRaRb, -(CH2)n-C(=Y)SRa, -OC(=Y)Ra, -OC(=Y)ORa, - OC(=Y)NRaRb, -OP(=0)RaRb, -(CH2)nOC(=Y)Ra , -(CH2)nOC(=Y)ORa, - (CH2)nOC(=Y)NRaRb, -(CH2)nOP(=0)RaRb, -N(Ra)C(=Y)Rb, -N(Ra)C(=Y)ORb, - N(R°)C(=Y)NRaRb, -NRaS(0)d Rb, -NHP(=0)RaRb, -(CH2)nNRaC(=Y)Rb, - (CH2)nNRaC(=Y)ORb, -(CH2)nNRcC(=Y)NRaRb, -(CH2)nNRaS(0)dRb or -
(CH2)nNHP(=0)RaRb; each of which may optionally be substituted at any available position by one or more substituents selected from Ci-12alkyl, C2-i2alkenyl, C2-i2alkynyl, C3.20cycloalkyl, heterocyclyl, aryl, heteroaryl, halogen , -CN, -N02 orNH2; or
R8 and R9 are joined together to form a monocyclic or polycyclic ring, which may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R1 1;
12 13
R and R are joined together to form a monocyclic or polycyclic ring, which may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R 1 1 ;
Rn is selected from hydrogen, halogen, Ci-]2alkyl, C2-i2alkenyl, C2_i2alkynyl, Ci_ i2haloalkyl, C2_i2 haloalkenyl, C2_i2haloalkynyl, Ci_|2alkoxy, Ci.]2haloalkoxy, C i_ 6alkoxyCi-6alkyl, Ci_6alkoxyCi.6alkoxyCi-3alkyl, Ci_i2alkylcarbonyl, C[_ i2alkoxycarbonyl, C3.2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, - (CH2)n-heterocyclyl, -(CH2),raryl, -(CH2)„-heteroaryl, -CN, -N02, -NRaRb, - (CH2)nNRaRb,-N3, -NCS, -(CH2)nN3, -(CH2)n NCS, -CRa (=NORb), -NRcNRaRb, -ORa, - SRa, -(CH^YR3, -S(0)dRa, -S(0)d NRaRb, -(CH2)nS(0)dRa, -(CH2)nS(0)dNRaRb, -
P(=0)RaRb, -(CH2)nP(=0) RaRb, -C(=Y)Ra, -C(=Y)ORa, -C(=Y)SRa, -C(=Y)NRaRb, - (CH2)nC(=Y)Ra, -(CH2)nC(=Y)ORa, -(CH2)nC(=Y)NRaRb, -(CH2)n-C(=Y)SRa, - OC(=Y)Ra, -OC(=Y)ORa, -OC(=Y)NRaRb, -OP(=0)RaRb, -(CH2)nOC(=Y)Ra , - (CH2)nOC(=Y)ORa, -(CH2)nOC(=Y)NRaRb, -(CH2)nOP(=0)RaRb, -N(Ra)C(=Y)Rb, - N(Ra)C(=Y)ORb, -N(R°)C(=Y)NRaRb, -NRaS(0)d Rb, -NHP(=0) RaRb, - (CH2)nNRaC(=Y)Rb, -(CH2)nNRaC(=Y)ORb, -(CH2)nNRcC(=Y)NRaRb, (CH2)nNRaS(0)dR or -(CH2)nNHP(=0) RaRb; each of which may optionally be substituted at any available position by one or more substituents selected from Q. i2alkyl, C2-i2alkenyl, C2.i2alkynyl, C3-20cycloalkyl, heterocyclyl, aryl, heteroaryl, , -CN, - N02 orNH2;
R16 is selected from hydrogen, CM2alkyl, C2-12alkenyl, C2-i2alkynyl, C3-20cycloalkyl, heterocyclyl, aryl, heteroaryl, -CRa(=NORb), -S(0)dRa, -S(0)d NRaRb, -(CH2)„S(0)dRa, - P(=0)RaRb, -C(=Y)Ra, -C(=Y)ORa, -C(=Y)SRa or -C(=Y)NRaRb, each of which may optionally be substituted at any available position by one or more substituents selected from Ci-i2alkyl, C2-i2alkenyl, C2-i2 alkynyl, C3-20cycloalkyl, heterocyclyl, aryl, heteroaryl, , -CN, -N02 orNH2;
Ra, Rb and Rc are independently selected from hydrogen, halogen, C|_i2alkyl, C2-i2alkenyl, C2-i2alkynyl, Ci-]2alkoxy, Ci- alkoxyCi-6alkyl, C1-12alkylcarbonyl, Ci.i2alkoxycarbonyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, -(CH2)n-heterocyclyl, -(CH2)n-aryl, -(CH2)n-heteroaryl, -CN, -N02, -N3, -NCS, -NR8R9, -(CH2)nNR8R9, - (CH2)nN3, -(CH2)nNCS, -CR8(=NOR9), -OH, -OR8, -CH2OH, -(CH2)nYR8 , - (C¾)nS(0)dR8, -(CH2)nS(0)dNR8R9, -(CH2)nP(=0)R8R9, -C(=Y)R8, -C(=Y)OR8, - C(=Y)SR8, -C(=Y)NR8R9, -(CH2)nC(=Y)R8, -(CH2)nC(=Y)OR8, -(CH2)nC(=Y)NR8R9, - (CH2)n-C(=Y)SR8, -OC(=Y)R8, -OC(=Y)OR8, -OC(=Y)NR8R9, -OP(=0)R8R9, - (CH2)nOC(=Y)R8 , -(CH2)nOC(=Y)OR8, -(CH2)n0C(=Y)NR8R9, -(CH2)nOP(=0)R8R9, - (CH2)nNR8C(=Y)R9, -(CH2)nNR8C(=Y)OR9, -(CH2)nNR10C(=Y)NR8R9, (CH2)nNR8S(0)dR9 or -(CH2)nNHP(=0)R8R9; each of which may optionally be substituted at any available position by one or more substituents selected from R1 1; wherein Ra and Rb can be joined together to form a monocyclic or polycyclic ring, which may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R1 ';
Re is selected from -cycloalkyl, -aryl, -heteroaryl, -YR8 , -C(=Y)R8, -C(=Y)OR8, - C(=Y)NR8R9, -C(=Y)SR8, -OC(=Y)R8 , -OC(=Y)OR8 or -OC(=Y)NR8R9 ; each of
which may optionally be substituted at any available position by one or more substituents selected from R11;
n is 0, 1, 2, 3, 4 or 5;
d is 1 or 2;
m is 1, 2, 3, 4 or 5.
A further aspect of the present invention provides processes for the preparation of the novel compounds of Formula I, their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
Another aspect of the present invention provides pharmaceutical compositions, containing compounds of Formula I, their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof in combination with one or more pharmaceutically acceptable carrier(s), adjuvants and vehicles.
Another aspect of the present invention is the use of the compounds of Formula I, their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, for the prophylaxis, amelioration and/or treatment of one or more condition(s)/disease(s)/ disorder(s), in a subject in need thereof.
Still another aspect of the present invention is the use of the compounds of Formula I, their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, for the prophylaxis, amelioration and/or treatment of one or more condition(s)/disease(s)/ disorder(s) that may be regulated or normalized via inhibition of SGLT-2.
Yet another aspect of the invention is to provide methods of using the compounds of Formula I of the present invention or compositions comprising the compounds of Formula I for the prophylaxis, amelioration and/or treatment of disease(s)/ disorder(s) involving SGLT-2 inhibition which comprises administering to a subject in need thereof the compounds of Formula I or compositions comprising a pharmaceutically effective amount of the compounds of Formula I.
A further aspect of the present invention is the use of a compound of Formula I for the manufacture of a medicament for the prophylaxis, amelioration and/or treatment of one or more condition(s)/disease(s)/ disorder(s) involving SGLT-2 inhibition in a subject in need thereof.
The present invention also encompasses prodrugs and active metabolites of the compounds of the Formula I.
0796
Other aspects of the invention will be set forth in the description which follows, and in part will be apparent from the description, or may be leamt by the practice of the invention. Detailed description of the invention
The present invention relates to the novel compounds of Formula I,

Formula I
their pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, wherein:
ring A represents aryl;
ring B represents either aryl or heteroaryl;
U, V and W are independently selected from -OH, hydrogen, halogen, C).]2alkoxy, -CN, -(CH2)„NR8R9, -OR8, -C(=Y)OR8 or -C(=Y)NR8R9; provided that atleast two groups out of U, V and W represent -OR8 ;
Y represents either O or S;
R7 is selected from halogen, Ci-i2alkyl, C2.i2alkenyl, C2-i2alkynyl, Ci-i2alkylcarbonyl, Ci-12 alkoxycarbonyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)nRe, -CN, -N02, - NR8R9, -N3, -CR8(=NOR9), -OH, -OR8, -CH2OH, -C(=Y)R8, -C(=Y)OR8, -C(=Y)SR8, - C(=Y)NR8R9, -OC(-Y)R8, -OC(=Y)OR8, -0C(=Y)NR8R9, -OP(=0)R8R9, -(CH2)„- heterocyclyl, -(CH2)n-NR8R9, -(CH2)n-N3, -(CH2)n-NCS, -(CH2)n-S(0)dR8, -(CH2)„- S(0)dNR8R9, -(CH2)„-P(=0)R8R9, -(CH2)„ -0P(=0)R8R9, -(CH2)a-NR8C(=Y)R9, -(CH2)n- NR8C(=Y)OR9, -(CH2)n-NR10C(=Y)NR8R9, -(CH2)„ -NR8S(0)dR9 or -(CH2)»- NHP(=0)R8R9 ; each of which may optionally be substituted at any available position by one or more substituents selected from R1 1 ;
R1, R2, R3 and R4 are independently selected from hydrogen, halogen, Ci_i2alkyl, C2-i2 alkenyl, C2_i2alkynyl, Ci-i2haloalkyl, C2-i2haloalkenyl, C2.i2haloalkynyl, Ci_i2alkoxy, C\. n haloalkoxy, Ci-6alkoxyCi-6alkyl, C]-6alkoxyCi.6alkoxyCi.3alkyl, Ci-i2alkylcarbonyl, C]-j2 alkoxycarbonyl, C3_2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, cycloalkenyl, cycloalkynyl, -(CH2)n-heterocyclyl, -(CH2)n-aryl, -(CH2)n-heteroaryl, -
CN, -NO2, -NR12R13, -(CH2)nNR1 R13, -N3, -NCO, -(CH2)nN3, -(CH2)n NCS, - CR12(=NOR13), -NR14NRI2R13, oxo, -OR12, -SR12, -(CH2)nYR12, -S(0)dR12, - S(0)dNR12R13, -(CH2)nS(0)dR12, -(CH2)nS(0)dNR,2R13, -P(=0)R12R13, - (CH2)nP(=0)R12R13, -C(=Y)R12, -C(=Y)OR12, -C(=Y)SR12, -C(=Y)NR12R13, - (CH2)nC(=Y)R12, -(CH2)nC(=Y)0R12, -(CH2)nC(=Y)NR12R13, -(CH2)n-C(=Y)SR12, - OC(=Y)R12, -OC(=Y)OR12, -OC(=Y)NR12R13,-OP(=0)R12R13, -(CH2)nOC(=Y)R12 , - (CH2)nOC(=Y)OR12, -(CH2)nOC(=Y)NR12R13, -(CH2)nOP(=0)R12R13, -N(R12)C(=Y)R13, -N(R1 )C(=Y)0R13, -N(R14)C(=Y)NR,2Ri3, -NR,2S(0)dR13, -NHP(=0)Ri2R!3, - (CH2)nNR, C(=Y)R13, -(CH2)nNRl2C(=Y)OR13, -(CH2)nNR14C(=Y)NR12R13, - (CH2)nNR12S(0)dR13 or -(CH2)nNHP(=0)R12R13; each of which may optionally be substituted at any available position by one or more substituents selected from Ru;
L is selected from O, S, SO, S02, -C(=0)-, -(CH2)n-, -C(=CH2)-, 1,1-cyclopropylene, - NR16- or -(C(R8)2)m- ; each methylene group may optionally be substituted with one or more substituents independently selected from halogen, hydroxy, oxo, -C(=0)0-, - C(=0)NR16-, Ci.i alkyl, C1-12alkoxy, -C3-20cycloalkyl or -C3.2ocycloalkoxy ;
E can be absent or is selected from CH2, O, S or NR16;
G can be absent or is selected from Ci-12alkylene, C2-i2alkenylene, C2-i2alkynylene, Ci-i2 alkylenecarbonyl, C3.20cycloalkylene, heterocyclyl, aryl, heteroaryl, -NR15-, - (CH2)nNR15-, -(CH2)nS(0)d-, -(CH2)nS(0)d NR15-, -(CH2)nP(=0)R15-, -C(=Y)-, C(=Y)NR15-, -(CH2)nC(=Y)-, -(CH2)nC(=Y)NR15-, -(CH2)nOC(=Y)- ,
(CH2)nOP(=0)R15- or -(CH2)nNR15S(0)d-; each of which may optionally be substituted at any available position by one or more substituents selected from R11;
R5 and R6 are independently selected from hydrogen, CM2alkyl, -S(0)dRa, -S(0)dNRaRb or -P(=0)RaRb; each of which may optionally be substituted at any available position by R1 ' ; wherein Ra and Rb can be joined together to form a monocyclic or polycyclic ring, which may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R1 1 ;
or
R5 and R6 are joined together along with the nitrogen atom to which they are attached to form a monocyclic or polycyclic ring, which contains atleast one phosphorus atom and may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by Rn;
provided that
(f) both R5 and R6 can not be hydrogen at the same time
(g) both R5 and R6 can not be alkyl at the same time
(h) R5 and R6 can not be a combination of hydrogen and alkyl at the same time
(i) when E and G are absent and R5 is hydrogen then R6 can not represent -S(0)dRa
(j) when R7 represents Cu2alkyl, C2-i2alkenyl, C2-i2alkynyl, -CH2OH or -(CH2)nRe, wherein n is not equal to zero; one of R5 and R6 represents -H or C1-6alkyl and the other represents -S(0)dRa, wherein d represents 1 or 2; then Ra can not be Ci. 6alkyl, C2- alkenyl, C2-6alkynyl, aryl or heteroaryl;
R8, R9, R10, R12, R13, R14 and R15 are independently selected from hydrogen, halogen, Q. i2alkyl, C2-i2alkenyl, C2-i2alkynyl, Ci-)2alkylcarbonyl, Ci-)2alkoxycarbonyl, C3- 2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, -(CH2)n-heterocyclyl, - (CH2)n-aryl, -(CH2)n-heteroaryl, -CN, -N02, -NRaRb, -(CH2)nNRaRb, -N3, -NCS, - (CH2)„N3, -(CH2)n NCS, -CRa(=NORb), -NRcNRaRb, -ORa, -SRa, -(CH2)nYRa, -S(0)dRa, -S(0)d NRaRb, -(CH2)nS(0)dRa, -(CH2)n S(0)d NRaRb, -P(=0)RaRb, -(CH2)n P(=0)RaRb, -C(=Y)Ra, -C(=Y)ORa, -C(=Y)SRa, -C(=Y)NRaRb, -(CH2)nC(=Y)Ra, - (CH2)nC(=Y)ORa, -(CH2)nC(=Y)NRaRb, -(CH2)n-C(=Y)SRa, -OC(=Y)Ra, -OC(=Y)ORa, - OC(=Y)NRaRb, -OP(=0)RaRb, -(CH2)nOC(=Y)Ra , -(CH2)nOC(=Y)ORa, - (CH2)nOC(=Y)NRaRb, -(CH2)nOP(=0)RaRb, -N(Ra)C(=Y)Rb, -N(Ra)C(=Y)ORb, - N(R°)C(=Y)NRaRb, -NRaS(0)d Rb, -NHP(=0)RaRb, -(CH2)nNRaC(=Y)Rb, - (CH2)nNRaC(=Y)ORb, -(CH2)nNR°C(=Y)NRaRb, -(CH2)nNRaS(0)dRb or -
(CH2)nNHP(=0)RaRb; each of which may optionally be substituted at any available position by one or more substituents selected from Ci-i2alkyl, C2_i2alkenyl, C2-i2alkynyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, halogen , -CN, -N02 orNH2; or
R8 and R9 are joined together to form a monocyclic or polycyclic ring, which may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R1 1;
R12 and R13 are joined together to form a monocyclic or polycyclic ring, which may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R1 1 ;
R1 1 is selected from hydrogen, halogen, Ci-i2alkyl, C2.i2alkenyl, C2-i2alkynyl, C|. i2haloalkyl, C2- i 2 haloalkenyl, C2-i2haloalkynyl, C].]2alkoxy, C].)2haloalkoxy, Cj.
6alkoxyCi. alkyl, Ci-6alkoxyCi-6alkoxyCi-3alkyl, Ci-i2alkylcarbonyl, Ci_ i2alkoxycarbonyl, C3-20cycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, - (CH2)n-heterocyclyl, -(CH2)n-aryl, -(CH2)n-heteroaryl, -CN, -N02, -NRaRb, - (CH2)nNRaRb,-N3, -NCS, -(CH2)nN3, -(CH2)n NCS, -CRa (=NORb), -NRcNRaRb, -ORa, - SRa, -(CH2)nYRa, -S(0)dRa, -S(0)d NRaRb, -(CH2)nS(0)dRa, -(CH2)„S(0)dNRaRb, - P(=0)RaRb, -(CH2)nP(=0) RaRb, -C(=Y)Ra, -C(=Y)ORa, -C(=Y)SRa, -C(=Y)NRaRb, - (CH2)nC(=Y)Ra, -(CH2)nC(=Y)ORa, -(CH2)nC(=Y)NRaRb, -(CH2)n-C(=Y)SRa, - OC(=Y)Ra, -OC(=Y)ORa, -OC(=Y)NRaRb, -OP(=0)RaRb, -(CH2)nOC(=Y)Ra , - (CH2)nOC(=Y)ORa, -(CH2)nOC(=Y)NRaRb, -(CH2)nOP(=0)RaRb, -N(Ra)C(=Y)Rb, - N(Ra)C(=Y)ORb, -N(R°)C(=Y)NRaRb, -NRaS(0)d Rb, -NHP(=0) RaRb, - (CH2)nNRaC(=Y)Rb, -(CH2)nNRaC(=Y)ORb, -(CH2)nNR°C(=Y)NRaRb, (CH2)nNRaS(0)dRb or -(CH2)„NHP(=0) RaRb; each of which may optionally be substituted at any available position by one or more substituents selected from Ci_ i2alkyl, C2-i2alkenyl, C2.j2alkynyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, , -CN, - N02 orNH2;
R16 is selected from hydrogen, Ci-12alkyl, C2-12alkenyl, C2-]2alkynyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -CRa(=NORb), -S(0)dRa, -S(0)d NRaRb, -(CH2)nS(0)dRa, - P(=0)RaRb, -C(=Y)Ra, -C(=Y)ORa, -C(=Y)SRa or -C(=Y)NRaRb, each of which may optionally be substituted at any available position by one or more substituents selected from Ci-i2alkyl, C2-i2alkenyl, C2-i2 alkynyl, C3.2ocycloalkyl, heterocyclyl, aryl, heteroaryl, , -CN, -N02 orNH2;
Ra, Rb and Rc are independently selected from hydrogen, halogen, Ci-i2alkyl, C2-12alkenyl, C2-i2alkynyl, Ci-12alkoxy, Ci.6alkoxyCi-6alkyl, Ci_i2alkylcarbonyl, Ci_i2alkoxycarbonyl, C3-20cycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, -(CH2)n-heterocyclyl, -(-CH2)n-aryl, -(CH2)n-heteroaryl, -CN, -N02, -N3, -NCS, -NR8R9, -(CH2)nNR8R9, - (CH2)11N3, -(CH2)nNCS, -CR8(=NOR9), -OH, -OR8, -CH2OH, -(CH2)nYR8 , - (CH2)nS(0)dR8, -(CH2)nS(0)dNR8R9, -(CH2)nP(=0)R8R9, -C(=Y)R8, -C(=Y)OR8, - C(=Y)SR8, -C(=Y)NR8R9, -(CH2)nC(=Y)R8, -(CH2)nC(=Y)OR8, -(CH2)nC(=Y)NR8R9, - (CH2)n-C(=Y)SR8, -OC(=Y)R8, -OC(=Y)OR8, -OC(=Y)NR8R9, -OP(=0)R8R9, - (CH2)„OC(=Y)R8 , -(CH2)nOC(=Y)OR8, -(CH2)nOC(-Y)NR8R9, -(CH2)nOP(=0)R8R9, -
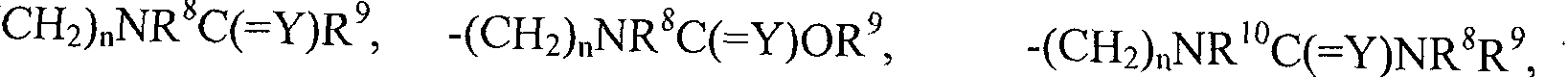
(CH2)nNR8S(0)dR9 or -(CH2)nNHP(=0)R8R9; each of which may optionally be substituted at any available position by one or more substituents selected from R1 1; wherein Ra and Rb can be joined together to form a monocyclic or polycyclic ring, which
may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by Ru;
Re is selected from -cycloalkyl, -aryl, -heteroaryl, -YR8 , -C(=Y)R8, -C(=Y)OR8, - C(=Y)NR8R9, -C(=Y)SR8, -OC(=Y)R8 , -OC(=Y)OR8 or -OC(=Y)NR8R9 ; each of which may optionally be substituted at any available position by one or more substituents selected from Ru;
n is 0, 1, 2, 3, 4 or 5;
d is 1 or 2;
m is 1, 2, 3, 4 or 5.
One embodiment of the present invention provides compounds of Formula la, wherein

Formula la
R1, R2, R3, R4, R5, R6, R7, U, V, W, E, G, ring A and ring B are as defined herein; their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
Another embodiment of the present invention provides compounds of Formula lb, wherein

Formula lb
R1, R2, R3, R4, R5, R6, R7, E, G, ring A and ring B are as defined herein; their
pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
Another embodiment of the present invention provides compounds of Fonnula Ic, wherein

Formula Ic
R1 , R2, R3, R4, R5, R6, R7, E and G are as defined herein; their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
Another embodiment of the present invention provides compounds of Formula Id, wherein

Formula Id
R5, R6, R7, E and G are as defined herein; their pharmaceutically acceptable derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
In another embodiment of the compounds of the present invention, it is preferred that E is selected from O or CH2.
In another embodiment of the compounds of the present invention, it is preferred that G is C1-12 alkylene which is unsubstituted or substituted at any available position by one or more substituents selected from Rn.
In still another embodiment of the compounds of the present invention, it is preferred that R7 is selected from the group consisting of -OR8, -(CH2)nYR8, - (CH2)nNR8R9, -(CH2)nNR10C(-Y)NR8R9, -(CH2)nOC(=Y)R8 and -(CH2)nOC(=Y)OR8, each of which is unsubstituted or substituted, at any available position, with one or more substituents selected from R11.
In a further embodiment of the compounds of the present invention, it is more preferred that R7 is selected from the group consisting of -OCH3, -CH2OH, - CH2OCH2CF3, -CH2OCOCH3, -CH2OCOC2H5, -CH2OCOC3H7, -CH2OCOC4H9, - CH2OCO(CH2)5CH3, -CH2OCO(CH2)7CH3, -CH2OCO(CH2)10CH3,
CH2OCO(CH2)14CH3, -CH2OCOCH2OCOCH3, -CH2OCOOCH3, -CH2OCOOC2H5, - CH2OCOOCH2CH(CH3)2, -CH2OCOOCH2C6H5,

each of which is unsubstituted or su st tute , at any ava a e pos t on, with one or more substituents selected from R 11.
In another embodiment of the compounds of the present invention, it is preferred that R5 and R6 are independently selected from the group consisting of -H, -CH3, -S02CH3,
-S02C2H5, -S02NH2, -S02N(CH3)2, -S02NHCOCH3, -S02-cycloalkyl, -S02-heterocyclyI, O o
II J II
¾-R— and V-R-0
\ 0 \
/
In still another embodiment of the compounds of the present invention, it is preferred that 5 and R6 together with the N atom to which they are attached represent

Definitions
Relative to the above description of the compounds of the present invention, the following definitions apply.
The term "alkyl" as used herein alone or as part of another group refers to a straight or branched chain aliphatic hydrocarbon chain, having from 1 to 12 carbon atoms. Examples of alkyl include, but are not limited to methyl, ethyl, n-propyl, isoprppyl, n-butyl, n-pentyl, t-butyl and the like. These groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
The term "alkenyl" as used herein alone or as part of another group refers to a straight or branched chain aliphatic hydrocarbon group containing at least one carbon- carbon double bond, having from 2 to 12 carbon atoms. Examples of alkenyl include, but are not limited to ethenyl, 1-propenyl, 2-propenyl, iso-propenyl, 1 -butenyl, 2-butenyl, and the like. These groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
The term "alkynyl" as used herein alone or as part of another group refers to a straight or branched chain aliphatic hydrocarbon group containing at least one carbon- carbon triple bond, having from 2 to 12 carbon atoms. Examples of alkynyl include, but are not limited to ethynyl, propynyl, and butynyl. These groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
The term "alkylene" as used herein refers to a divalent straight or branched chain aliphatic hydrocarbon group, having from 1 to 12 carbon atoms. Examples of alkylene include, but are not limited to methylene, ethylene, isopropylene, n-butylene, 1 ,1 - dimethylethylene and the like. These groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
The term "alkenylene" as used herein refers to a divalent straight or branched chain aliphatic hydrocarbon group containing at least one carbon-carbon double bond, having from 2 to 12 carbon atoms. Examples of alkenyl include, but are not limited to ethenylene, 1 -propenylene, 2-propenylene, iso-propenylene, 1 -butenylene, 2-butenylene, and the like. These groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
The term "alkynylene" as used herein refers to a divalent straight or branched chain aliphatic hydrocarbon group containing at least one carbon-carbon triple bond, having from 2 to 12 carbon atoms. Examples of alkynyl include, but are not limited to ethynylene, propynylene, and butynylene. These groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, cyano, amino, nitro, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
The term "alkoxy" refers to an above defined alkyl group attached via an oxygen linkage to the rest of the molecule. Non-limiting examples of such groups include -OCH3, -OC2¾ and the like.
The term "alkoxyalkyl" refers to an above defined alkyl group, in which one or more hydrogen atoms are replaced by alkoxy group as defined herein. Non-limiting examples include -CH2OCH3, -CH2OC2H5, -CH2CH2OC2H5 and the like.
The term "alkoxyalkoxyalkyl" refers to an above defined alkoxyalkyl group, in which one or more hydrogen atoms are replaced by above defined alkoxy group. Non- limiting examples include -CH2OCH2OCH3, -CH2OCH2CH2OC2H5, CH2CH2OCH2OC2H5 and the like.
The term "alkyl carbonyl" refers to an above defined alkyl group attached via a carbonyl linkage to the rest of the molecule. Non-limiting examples of such groups include -C(0)CH3, -C(0)C2H5 and the like.
The term "alkoxycarbonyl" refers to an above defined alkoxy group attached via a carbonyl linkage to the rest of the molecule. , Non-limiting examples of such groups include -C(0)-0 CH3, -C(0)-OC2H5, and the like.
The term "halogen" refers to F, CI, Br or I.
The term "haloalkyl" refers to an above-defined "alkyl" group, which is substituted with one or more "halogen" groups, as defined herein, at any one or more of the 1 to 12 carbon atoms of the alkyl group. Representative examples of haloalkyl include, but are not limited to, chloromethyl, fluoromethyl, trifluoromethyl, trichloromethyl, difluoroethyl, trifluoroethyl, dichloroethyl, and the like.
The term "haloalkenyl" refers to an above-defined "alkenyl" group, which is substituted with one or more "halogen" groups, as defined herein, at any one or more of the carbon atoms of the alkenyl group. Representative examples of haloalkenyl include, but are not limited to, chloroethenyl, 2-fluroethenyl, triflurobutenyl, dichloropropenyl and the like.
The term "haloalkynyl" refers to an above-defined "alkynyl" group, which is substituted with one or more "halogen" groups, as defined herein, at any one or more of the carbon atoms of the alkynyl group. Representative examples of haloalkynyl include, but are not limited to, 2-fluroethynyl, triflurobutynyl, dichloropropynyl and the like.
The term "haloalkoxy" refers to an above defined "haloalkyl" group, appended to the parent molecular moiety through an oxygen atom.
The term "cycloalkyl" refers to cyclic alkyl groups consisting of 3 to 20 carbon atoms having a single cyclic ring or multiple condensed rings, for example, fused or spiro systems, which may be partially unsaturated, unless otherwise constrained by the definition. Such cycloalkyl groups include, by way of example, single ring structures, for
example, cyclopropyl, cyclobutyl, cyclopentenyl, cyclohexyl, cyclooctyl, and the like, or multiple ring structures, for example, adamantyl, and bicyclo[2.2.1] heptane, or cyclic alkyl groups to which is fused an aryl group, for example, indane and the like. Cycloalkyl groups may further be substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, carboxyalkyl, azido, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
The term "aryl" herein refers to a mono- or poly- carbocyclic aromatic group, for example phenyl or naphthyl ring and the like optionally substituted with one or more substituents selected from but not limited to, for example, halogen, hydroxy, alkyl, alkenyl, alkynyl, cycloalkyl, alkoxy, acyl, amino, aryloxy, CF3, COORd (wherein Rd can be hydrogen, alkyl, alkenyl, cycloalkyl, aralkyl, heterocyclylalkyl or heteroarylalkyl), cyano, nitro, carboxy, heterocyclyl, heteroaryl, heterocyclylalkyl or heteroarylalkyl. The aryl group may optionally be fused with cycloalkyl group, heteroaryl group, heterocyclyl group or another aryl group. The fused group may be further substituted at any available position with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, amino, nitro, cyano, carboxyalkyl, azido, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl, acyloxy, aryl or heterocyclyl, heteroaryl.
The term "aryloxy" refers to an above defined aryl group attached via an oxygen linkage to the rest of the molecule, for example -OPh and the like.
The term "heteroaryl" unless and otherwise specified refers to an aromatic monocyclic or polycyclic ring structure, containing one or more heteroatoms independently selected from N, O, S or P. "Heteroaryl" also includes, but is not limited to, bicyclic or tricyclic rings, wherein the heteroaryl ring is fused to one or two rings independently selected from the group consisting of an aryl ring, a cycloalkyl ring, a heterocyclyl ring and another monocyclic heteroaryl ring. Examples of heteroaryl groups include, but not limited to, oxazolyl, imidazolyl, pyrrolyl, 1 ,2,3-triazolyl, 1,2,4-triazolyl, tetrazolyl, thiazolyl, ox_adiazolyl, benzoimidazolyl, thiadiazolyl, pyridinyl, pyridazinyl, pyrimidinyl, thienyl, isoxazolyl, triazinyl, furanyl, benzofuranyl, indolyl, benzothiazolyl, benzoxazolyl, imidazo[l,2-a]pyrimidine, imidazo[l ,2-a]pyrazine, and the like. The bicyclic or tricyclic heteroaryl rings can be attached either through the heteroaryl group itself or the aryl, cycloalkyl or heterocyclyl group to which it is fused. The heteroaryl group may be further substituted at any available position with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, amino, nitro, cyano,
carboxyalkyl, azido, alkenyl, alkynyl, alkoxy, cycloalkyl, cycloalkynyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
The term "heterocyclyl" unless otherwise specified refers to a non-aromatic monocyclic or polycyclic cycloalkyl group, for example, fused or spiro systems fully or partially unsaturated, containing one or more heteroatom(s) independently selected from N, O, S or P. The nitrogen, sulphur and phosphorus heteroatoms may optionally be oxidized. The nitrogen atoms may optionally be quaternerized. The heterocyclyl ring may be fused with another cycloalkyl, aryl, heterocyclyl or heteroaryl ring and are optionally benzofused or fused heteroaryl of 5-6 ring members and/or are optionally substituted wherein the substituents are selected from but not limited to halogen, hydroxy, alkyl, alkenyl, alkynyl, cycloalkyl, acyl, carboxy, aryl, alkoxy, aralkyl, cyano, nitro, amino, heterocyclyl, or heteroaryl. Examples of heterocyclyl groups include but are not limited to, morpholinyl, oxazolidinyl, tetrahydrofuranyl, dihydrofuranyl, dihydropyridinyl, dihydroisooxazolyl, dihydrobenzofuryl, azabicyclohexyl, dihydroindonyl, piperidinyl or piperazinyl. The fused group may be further substituted at any available position with one or more substituents selected from but not limited to, for example, halogen, hydroxy, oxo, carboxy, amino, nitro, cyano, carboxyalkyl, azido, alkenyl, alkynyl, alkoxy, cycloalkyl, acyl acyloxy, aryl, heterocyclyl or heteroaryl.
The term "hydroxy" refers to the group -OH.
The term "oxo" refers to carbonyl group represented as >C=0.
In all the above definitions, nitrogen, sulphur and phosphorus heteroatom can optionally be quaternerized or oxidized wherever possible.
The term "Protecting Group" or "PG" refers to a group which is in a modified form to preclude undesired side reactions at the protected site. The term protecting group, unless otherwise specified, may be used with groups, for example, hydroxy, amino, carboxy and examples of such groups are found in T.W. Greene, et al. "Protecting Groups in Organic Synthesis " 3ld Ed, Wiley, New York, which is incorporated herein by reference. The species of the carboxylic protecting groups, amino protecting groups or hydroxy protecting groups employed are not critical, as long as the derivatised moieties/moiety is/are stable to conditions of subsequent reactions and can be removed without disrupting the remainder of the molecule. Examples of suitable hydroxy and amino protecting groups include but are not limited to trimethylsilyl, triethylsilyl, o-nitrobenzyloxycarbonyl, p- nitrobenzyloxycarbonyl, f-butyldiphenylsilyl, i-butyldimethylsilyl, acetyl, trifluoroacetyl, benzyloxycarbonyl (CBz), t-butoxycarbonyl (Boc), 9-fluorenylnethylenoxycarbonyl
(Fmoc), 2,2,2-trichloroethyloxycarbonyl, allyloxycarbonyl and the like. Examples of suitable carboxy protecting groups are benzhydryl, o-nitrobenzyl, -nitrobenzyl, 2- naphthylmethyl, allyl, 2-chloroallyl, benzyl, 2,2,2- trichloroethyl, trimethylsilyl, t- butyldimethylsilyl, t-butyldiphenylsilyl, 2-(trimethylsilyl)ethyl, phenacyl, p- methoxybenzyl, acetonyl,/?-methoxyphenyl, 4-pyridylmethyl, -butyl and the like.
"Subject" includes humans, non-human mammals (e.g., dogs, cats, rabbits, cattle, horses, sheep and the like) or non-mammals (e.g., birds and the like).
The term "therapeutically effective amount" means the amount of a compound that, when administered to a subject for treating a disease, is sufficient to effect such treatment for the disease. The "therapeutically effective amount" will vary depending on the compound, the disease and its severity, weight, physical condition and responsiveness of the subject to be treated, among other factors. -.
A "pharmaceutically acceptable salt" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids. A "pharmaceutically acceptable salt" also encompasses any compound according to the present invention that is utilized in the form of a salt thereof.
Asymmetric centres may exist in the compounds of the present invention. The compounds of Formula I may have one or more stereogenic centres and so can exhibit optical isomerism. All such isomers including enantiomers, diastereomers, and epimers are included within the scope of this invention. Furthermore, the invention includes such compounds as single isomers (R and /or S) and as mixtures, including racemates. If desired, racemic mixtures of the compounds may be separated so that the individual enantiomers are isolated. The separation may be carried out by methods well known in the art, such as the coupling of a racemic mixture of compounds to an enantiomerically pure compound to form a diastereomeric mixture, followed by separation of the individual diastereomers by standard methods, such as fractional crystallization or chromatography. Starting materials of particular stereochemistry may either be commercially available or may be made by the methods described herein and resolved by techniques well known in the art. The independent syntheses of these diastereomers or their chromatographic separations may be achieved as known in the art by appropriate modifications.
Certain compounds according to Formula I, can also exist as tautomers, which have different points of attachment of hydrogen accompanied by one or more double bond
shifts. These tautomers, either separately or as mixtures, are also considered to be within the scope of the invention.
Certain compounds according to Formula I, may also exist as polymorphs.
The present invention also encompasses geometrical isomers of compounds of 5 Formula I and the mixtures thereof. The geometrical isomers may exist in E or Z; Syn or anti configurations. These geometrical isomers, either separately or as mixtures, are also considered to be within the scope of the invention.
Particularly useful examples of the present invention include but are not limited to the compounds selected from Table 1, including their pharmaceutically acceptable 10 derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof:
Table 1



The compounds disclosed herein may be prepared by techniques well known in the art and familiar to the skilled organic chemist. The compounds of the present invention may be prepared by the following reaction sequences as depicted in for example Scheme No 1 to 5. All of the starting materials are either commercially available or can be prepared by procedures that would be well known to one of ordinary skill in organic chemistry.
"Lg" is used to denote an appropriate leaving group and as such may vary in nature depending on the exact reaction conditions employed. Some typical leaving groups may be fluoro, chloro, bromo, iodo, tosyl, mesyl, trifluoromethanesulfonyl and the like, but these should not be construed as limiting as many other leaving groups are also well known to those skilled in the art.
Scheme 1

Formula I
Formula II
The compounds of the Formula I can be prepared from the compounds of Formula II following the steps provided in Scheme 2 or Scheme 3.
Scheme 2

The compounds of the Formula II can be coupled with compounds of Formula III to obtain compounds of Formula IV. The compounds of Formula III contain a N- protection group (PG) such as but not limited to ter -butoxycarbonyl, Fluorenyloxycarbonyl, Allyloxycarbonyl or Benzyloxycarbonyl etc. and a leaving group (Lg) for example but not limited to a halogen, triflate, mesylate, tosylate or boronate and the like. The procedures to couple Formula II with Formula III include but not limited to, mitsunobu reaction involving reagents such as diethylazadicaorboxylate, diisopropylazadicarboxylate in presence of triphenylphosphene in a suitable solvent such as but not limited to THF, Dioxane, DCM and the like or catalytic reactions involving Pd, Cu etc metal derivatives in presence or absence of a ligand or procedures , involving treatment of Formula II with Formula III in presence of a base for example but not limited
to NaH, KH, Cs2C0 , K2C03, NEt3, pyridine and the like in a suitable solvent such as THF, Dioxane, DMF, DMSO, acetone, dichloroethane, DCM or combination there of. The protection group from the compounds of Formula IV can be released by methods known to a person skilled in the art. Such deprotection methods include but not limited to, treatment with acids such as trifluoro acetic acid, HCl, H2SO4, HBr, HI etc.in a suitable solvent like DCM, Dichloroethane, diethylether, diisopropylether, THF, dioxane, actonitrile etc. or combination there of. Another type of deprotection methods include catalytic reductive methods involving Pd/C in presence of hydrogen in a suitable solvent such as but not limited to ethylacetate, methanol, acetic acid and the like or combination thereof. The compounds of Formula IV after deprotection can be converted to Formula I by treatement with compounds of Formula R6-Lg in presence of bases such as, but not limited to triethylamine, N-ethyldiisopropylamine, pyridine and the like in suitable solvents such as DCM, dichloroethane, THF, acetonitrile or combination thereof.
Scheme 3

Formula I
Alternately, the compounds of the Formula II, wherein Q represents particularly, - Ci-i2alkyl-OH can be reacted with reagents such as but not limited to, methanesulfonylchloride, p-toluenesulfonylchloride, trifuorosulfonicanhydride, thionylchloride, phosphorousoxychloride, carbontetrachloride, borontribromide, phosphoroustribromide, carbontetrabromide, iodine etc. in presence or absence of bases such as but not limited to triethylamine, N-ethyldiisopropylamine, pyridine, imidazole, triphenylphosphene in suitable solvents such as DCM, dichloroethane, THF, acetonitrile, carbontetrachloride to convert into compounds of Formula V. The compounds of Formula V can be converted to compounds of Formula VI by treating with sodiumazide in solvents such as but not limited to DMF, DMSO etc. The compounds of Formula VI can be converted to compounds of Formula VII by methods known to a person skilled in the art. Such methods include but not limited to treatment with triphenylphosphene in solvent like THF in presence of water or reduction in hydrogen atmosphere in presence of Pd/C to afford compounds of Formula VII. The compounds of Formula VII can be converted to Formula I by treatment with compounds of Formula R6-Lg in presence of bases such as, but not limited to triethylamine, JV-ethyldiisopropylamine, pyridine etc. in suitable solvents like DCM, dichloroethane, THF, acetonitrile or combination thereof.
Scheme 4

X is halogen
-OPG, -SPG, -N(PG)R16, -C^^alkyl-OPG
Formula IX

-C 2alkyl-OH
Formula II
The compounds of the Formula II can be prepared from Formula VIII and compounds of Formula IX by following Scheme 4. Formula IX can be prepared by following the procedure given in NUCLEOSIDE, NUCLOETIDES & NUCLEIC ACIDS, 20(4-7), 649-652 (2001). The compounds of Formula VIII can be reacted with metalated species generated by treating Formula IX (prepared according to the procedure given in US20070049537) with reagents such as but not limited to, "BuLi, 'BuLi or 'PrMgCl in presence or absence of LiCl etc. in a suitable solvent such as but not limited to, THF, diiethylether or combination thereof to obtain the compounds of Formula X. The compounds of Formula X can be converted to the compounds of Formula II by deprotection methods kown to a person skilled in the art. These deprotection methods include but not limited to, treatment with acids such as trifluoro acetic acid, HC1, H2S04, HBr, HI etc.in a suitable solvent like DCM, Dichloroethane, diethylether, diisopropylether, THF, dioxane, actonitrile etc. or combination there of.
eme 5

X is halogen
Q-PG is -OPG, -SPG, -N(PG) 16, -C^^alkyl-OPG
Formula IX

Formula XI N(PG)R16, -C1-12alkyl-OPG
Q-PG is -OPG, -SPG, -N(PG)R16, -C-,.12alkyl-OPG
Formula XII M = Metal
Reduction

Formula II N(PG)R16, -CL^alkyl-OPG
Formula XIII
Alternately, the compounds of the Formula 11 can be prepared by the steps provided in Scheme 5. Formula XI can be prepared by following the procedure given in US20070049537. Formula XI can be reacted with metalated species generated by treating Formula IX (prepared according to the procedure given in US20070049537) with reagents such as but not limited to, "BuLi, 'BuLi or 'PrMgCl in presence or absence of LiCl and the like in a suitable solvent such as but not limited to, THF, diiethylether or combination thereof. This step is followed by a suitable acetal formation reaction, for example but not limited to, treatment with methanesulfonicacid in presence of methanol to obtain compounds of Formula XII. The compounds of Formula XII can be reduced by treating with triethylsilane in presence of borotri fluoride etherate in acetonitrile to furnish the compounds of Formula XIII. The compounds of Formula XIII can be converted to the compounds of Formula II by deprotection methods kown to a person skilled in the art.
These deprotection methods include but not limited to, treatment with acids such as trifluoro acetic acid, HC1, H2S04j HBr, HI and the like in a suitable solvent like DCM, Dichloroethane, diethylether, diisopropylether, THF, dioxane, actonitrile and the like or combination there of. Alternately, compounds of Formula XIII can be converted to compounds of Formula II by deprotection reactions involving reagents such as but not limited to tetrabutylammoniumfluoride or Hydrogenfluoride in solvents such as DCM, dichloroethane, THF, dioxane or pyridine and the like.
It is understood that, as used herein, references to the compounds of structural Formula I are meant to also include the pharmaceutically acceptable salts, and also salts that are not pharmaceutically acceptable when they are used as precursors to the free compounds or their pharmaceutically acceptable salts or in other synthetic manipulations. The compounds of the present invention may be administered in the form of a pharmaceutically acceptable salt. The term "pharmaceutically acceptable salt" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids. The salts may be prepared during the final isolation and purification of the compounds or separately by making basic or acidic addition salts. Representative salts of basic compounds of the present invention can be prepared by reacting free base form of the compound with a suitable acid, including, but not limited to acetate, trifluoroacetate, adipate, citrate, aspartate, benzoate, benzenesulphonate, bisulfate, besylate, butyrate, camphorsulphonate, difluconate, hemisulfate, heptanoate, formate, fumarate, lactate, maleate, methanesulfonate, naphthylsulfonate, nicotinate, oxalate, picrate, pivalate, succinate, tartrate, tirchloracetat, glutamate, >-toluenesulphonate, hydrochloric, hydrobromic, sulphuric, phosphoric and the like. Representative salts of acidic compounds of the present invention can be prepared by reacting free acid form of the compound with a suitable base, including, but not limited to ammonium, calcium, magnesium, potassium, sodium salts, salts of primary, secondary and tertiary amines, substituted amines including naturally occurring ones e.g., arginine, betaine, caffeine, choline, glucamine, glucosamine, histidine, lysine, morpholine, piperazine, piperidine, purine, triethylamine and the like. Compounds of the present invention that contain a carboxylic acid (- COOH) or alcohol group, their pharmaceutically acceptable esters of carboxylic acids such as methyl, ethyl and the like, or acyl derivatives of alcohols such as acetate and the like, can be employed. Compounds of the present invention that comprise basic nitrogen atom may be quaternized with alkyl halides, alkyl sulfates and the like. Such salts permit
the preparation of both water soluble and oil soluble compounds of the present invention. It should be recognized that the free base or free acid forms will typically differ from their respective salt forms somewhat in physical properties such as solubility in polar solvents, but otherwise the salts are equivalent to their respective free forms for the purpose of the invention.
The "pharmaceutically acceptable solvates" refer to solvates with water (i.e., hydrates) or pharmaceutically acceptable solvents, for example, ethanol and the like.
The invention also encompasses "prodrugs" of the compounds of the present invention which upon in-vivo administration undergo cleavage by metabolic processes before becoming active pharmacological substances. In general such prodrugs are derivatives of functional group of a compound of the invention which are readily convertible in vivo into the compound of the invention. Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in "Targeted prodrug design to optimize drug delivery", AAPS PharmaSci (2000), 2(1), E6.
The invention also encompasses active "metabolites" of the compound of the present invention. An active metabolite is an active derivative of a SGLT-2 inhibitor produced when the SGLT-2 inhibitor is metabolized.
Various "polymorphs" of a compound of general Formula I forming part of this invention may be prepared by crystallization of a compound of Formula I under different conditions. For example, by using different solvents commonly used or their mixtures for recrystallization; crystallizations at different temperatures; various modes of cooling, ranging from very fast to very slow cooling during crystallizations, heating or melting the compound followed by gradual or fast cooling may also obtain polymorphs. The presence of polymorphs may be determined by solid probe NMR spectroscopy, IR spectroscopy, differential scanning calorimetry, powder X-ray diffraction or such other techniques.
The present invention includes all pharmaceutically acceptable isotopically- labeled compounds of Formula I wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes suitable for inclusion in the compounds of the present invention comprises isotopes of hydrogen, such as 2H and 3H, carbon, such as "C, 13C and l4C, chlorine, such as 36C1, fluorine, such as F, iodine, such as I and I, nitrogen, such as N and N, oxygen, such as O,
1 7 1 8 32 ^ 5
O and O, phosphorus, such as P, and sulphur, such as S. Substitution with heavier
isotopes such as deuterium, i.e. 2H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances. Isotopically- labeled compounds of Formula I can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying examples and schemes using an appropriate isotopically-labeled reagent in place of the non-labeled reagent previously employed.
The present invention also includes all the intermediate complexes of the compounds of Formula I, which are active by themselves or can be readily converted to compounds having inhibitory effect on sodium-dependent glucose cotransporter (SGLT), preferably SGLT-2.
The present invention also provides pharmaceutical compositions, comprising compounds of general Formula I or their pharmaceutically acceptable analogs, derivatives, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof together with one or more pharmaceutically acceptable carriers comprising excipients and auxiliaries, which facilitate processing of the active compound into preparations, which can be used pharmaceutically. The pharmaceutical compositions may be in the forms normally employed, such as tablets, capsules, powders, syrups, solutions, suspensions, emulsions, pills, granules, suppositories, pellets, depot formulations and the like, may contain flavourants, sweeteners etc in suitable solid or liquid carriers or diluents, or in suitable sterile media to form injectable solutions or suspensions. Such compositions typically contain from 0.1 to 99.9 % by weight of active compound, the remainder of the composition being pharmaceutically acceptable carriers, diluents or solvents.
The pharmaceutical compositions of the present invention can be manufactured by the processes well known in the art, for example, by means of conventional mixing, dissolving, dry granulation, wet granulation, dragee-making, levigating, emulsifying, encapsulating, entrapping, lyophilizing processes or spray drying. The compounds or the pharmaceutical compositions comprising such compounds of the present invention may be administered in the form of any pharmaceutical formulation. The pharmaceutical formulation will depend upon the nature of the active compound and its route of administration. Any route of administration may be used, for example oral, buccal, pulmonary, topical, parenteral (including subcutaneous, intramuscular and intravenous), transdermal, ocular (ophthalmic), by inhalation, intranasal, transmucosal, implant or rectal
administration. Preferably the compounds of the present invention are administered orally, parenterally or topically.
In an embodiment, the amount of the novel compounds having the Formula I according to the present invention to be incorporated into the pharmaceutical compositions of the present invention can vary over a wide range depending on known factors such as, for example, the disorder to be treated, the severity of the disorder, the patient's body weight, the dosage form, the chosen route of administration and the number of administration per day. Typically, the amount of the compound of Formula I in the pharmaceutical compositions of the present invention will range from approximately 0.01 mg to about 5000 mg. In an embodiment, the daily dose of composition comprising the novel compounds having the Formula I is in the range of about 0.01 mg/kg to about 100 mg/kg based on the body weight of the subject in need thereof which may be administered as a single or multiple doses.
In an embodiment, the novel compounds having the Formula I according to the present invention are particularly useful for the treatment of disease(s) or disorder(s), which are chronic or acute in nature, which favorably respond to or are alleviated by the novel compounds having the Formula I or compositions comprising them. The compositions comprising the novel compounds having the Formula I are useful prophylactically or therapeutically depending upon the pathological condition intended to be prevented or treated respectively.
In one embodiment compounds of the present invention are useful in the prophylaxis, amelioration and/or treatment of one or more condition(s)/disease(s)/ disorder(s), in a subject in need thereof.
In another embodiment compounds of the present invention are useful in the prophylaxis, amelioration and/or treatment of one or more condition(s)/disease(s)/ disorder(s), which may be regulated or normalized via inhibition of Sodium Glucose Cotransporters (SGLT).
The compounds of the present invention possess activity as selective inhibitors of SGLT-2 and are therefore useful for the prophylaxis, amelioration and/or treatment of variety of diseases, disorders and conditions, including, but not limited to, diabetes (including Type I and Type II), Metabolic Syndrome or 'Syndrome X' including impaired glucose tolerance, insulin resistance, metabolic acidosis or ketosis, disorders of food intake, satiety disorders, obesity, hyperinsulinemia, dyslipidemia (including hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels, high LDL
levels), hypertension associated with metabolic disorders, congestive heart failure, edema, hyperuricemia, gout, wound healing and tissue ischemia.
The compounds of the present invention can also be used for the prophylaxis, amelioration and/or treatment of the diseases, disorders and conditions collectively referenced to as "diabetic complications" which include both acute complications and chronic complications. Examples of "acute complications" include hyperglycemia (e.g., ketoacidosis), infections (e.g., skin, soft tissue, biliary system, respiratory system and urinary tract infections), etc. Examples of "chronic complications" include microangiopathy (e.g., nephropathy, retinopathy), arteriosclerosis (e.g., atherosclerosis, heart infarction, brian infarction, lower extremity arterial occlusion), neuropathy (e.g, sensory nerves, motor nerves, autonomic nerves), foot gangrene, etc. Major diabetic complications include diabetic retinopathy, diabetic nephropathy and diabetic neuropathy.
All of the various forms and sub-forms of the disorders mentioned herein are contemplated as part of the present invention.
A further embodiment of the present invention is the use of a compound of Formula
I for the manufacture of a medicament for the prophylaxis, amelioration and/or treatment of one or more condition(s)/ disease(s)/ disorder(s) involving SGLT-2 inhibition in a subject in need thereof.
Another embodiment of the present invention provides methods for the prophylaxis, amelioration and/or treatment of one or more condition(s)/ disease(s)/ disorder(s) involving SGLT-2 inhibition in a subject in need thereof that comprises administering a therapeutically effective amount of compound of Formula I.
In still another embodiment of the present invention is provided use of the dosage form compositions comprising the novel compounds of Formula I for the treatment of one or more condition(s)/ disease(s)/ disorder(s) involving SGLT-2 inhibition which comprises administrating to a subject in need thereof a pharmaceutically effective amount of the composition.
An embodiment of the present invention relates to methods of using the compounds of Formula I of the present invention or compositions comprising the compounds of Formula I for the prophylaxis, amelioration and/or treatment of any one or more condition(s)/ disease(s)/ disorder(s), which comprises administering to a subject in need thereof the compounds of Formula I or compositions comprising a pharmaceutically effective amount of the compounds of Formula I.
In yet another embodiment, the compounds or their pharmaceutically acceptable salts according to the present invention are useful in the treatment of the aforementioned diseases, disorders and conditions in combination with at least one other therapeutic agent. The compounds of the present invention may be used in combination with one or more other therapeutic agents in the treatment, prevention, suppression or amelioration of diseases or conditions for which compounds of the present invention or other therapeutic agents may have utility, where the combination of drugs together are safer or more effective than either drug alone.
Other therapeutic agents suitable for combination with the compounds of the present invention include, but are not limited to, known therapeutic agents useful in the treatment of the aforementioned disorders including: anti-diabetic agents; agents for prevention of complications of diabetes; anti-hyperglycemic agents; hypolipidemic/ lipid lowering agents; anti-obesity agents; anti-hypertensive agents, anti-platelet agents, anti- atherosclerotic agents, an ti -inflammatory agents, uricosuric agents, anti-TNF agent or c- AMP raising agents and appetite suppressants.
It is believed that the use of the compounds of the present invention in combination with atleast one or more of the aforementioned other therapeutic agents may provide results greater than that possible from each of these medicaments alone or greater than the combined additive effects produced by these medicaments. The present compounds and the other therapeutic agents may be administered in the same dosage form or in a separate dosage form by same or different administration route, in dosages and regimens as generally known in the art. Those agents which potentiate the therapeutic effect of SGLT- 2 inhibitors according to the invention may allow the dosage to be reduced.
Examples of suitable anti-diabetic agents for use in combination with the compounds of the present invention include but are not limited to (a) other SGLT-2 inhibitors; (b) insulin sensitizers including (i) PPAR γ agonists such as thiozolidinediones or glitazones (e.g. pioglitazone, rosiglitazone and the like), PPAR δ agonists, PPAR a agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), PPARpan agonists, PPAR γ/δ agonists, PPAR α/γ dual agonists, PPAR α/δ dual agonists, PPAR γ antagonists, PPAR α/γ modulators and PPAR α/γ/δ modulators, (ii) biguanides such as metformin and phenformin, and (iii) protein tyrosine phosphatase- IB (PTP-IB) inhibitors; (c) insulin or insulin mimetics; (d) sulfonylureas and other insulin secretagogues, such as tolbutamide, chlorpropamide,
tolazamide, glyburide (glibenclamide), glipizide, gliclazide, gliquidone, glimepiride, and meglitinides, such as repaglinide, mitiglinide, nateglinde and the like; (e) glucose absorption inhibitors like alpha.-glucosidase inhibitors (such as acarbose, voglibose and miglitol); (f) glucagon receptor antagonists; (g) GLP-1, GLP-1 mimetics such as exendin- 4 or amylin and GLP-1 receptor agonists (h) GIP and GIP mimetics (i) PACAP, PACAP mimetics, and PACAP receptor agonists; (j) AMPK activators; (k) Ι ΐβ-HSD inhibitors; (1) DPP-IV inhibitors such as Sitagliptin(Merck), Vildagliptin (Novartis); (m) inhibitors of glucose-6-phosphate, fructose- ,6-biphosphate, glycogen phosphorylase, phosphoenol pyruvate carboxykinase, glycogen synthase kinase, aminopeptidase-N or pyruvate dehydrokinase; (n) glucokinase activators (GKAs); (o) RXR modulators; (p) GPR40 agonists/antagonists, GPR1 19 agonists or GPR120 agonists; (q) alpha2 -antagonists; (r) IBAT inhibitors, HM74a/HM74 agonists, glucocorticoid antagonists, amylin receptor agonists, peptide YY hormone, PEPCK inhibitor, somatotropin release inhibiting factor, CPT-1 inhibitor, insulin receptor kinase stimulants, tripeptidyl peptidase II inhibitors, hepatic gluconeogenesis inhibitors or carboxypeptidase inhibitor.
Examples of suitable agents to be used in combination with the compounds of the present invention, for treatment or prevention of complications of diabetes include but are not limited to GABA-receptor antagonists, Na-channel blockers (e.g. mexiletine hydrochloride, oxacarbazepine or the like), γ-aminobutyric acid receptor antagonists (e.g. topiramat or the like), protein-kinase C inhibitors (e.g. midostaurin or the like), advanced glycation end product inhibitors (e.g. pyridoxamine or the like), transcript factor NF-KB inhibitors (e.g. dexlipotam or the like), lipid peroxide inhibitors (e.g. tirilazad mesylate or the like), a-linked-acid-dipeptidase inhibitors, carnitine derivatives (e.g. levacecamine, levocarnitine or the like), insulin like growth factor-I, platelet-derived growth factor, platelet-derived growth factor analogues, epidermal growth factor, nerve growth factor, biclomol, sulodexide or aldose reductase inhibitors (e.g. ascorbyl gamolenate, tolrestat, epalrestat or the like).
Examples of suitable hypolipidemic/ lipid lowering agents for use in combination with the compounds of the present invention include but are not limited to (a) cholesterol lowering agents such as (i) HMG-CoA reductase inhibitors (lovastatin, simvastatin, pravastatin, cerivastatin, fluvastatin, atorvastatin, itavastatin, and rosuvastatin, and other statins), (ii) bile acid sequestrants (cholestyramine, colestipol, and dialkylaminoalkyl derivatives of a cross-linked dextran), (iii) nicotinyl alcohol, nicotinic acid or a salt
thereof, (iv) PPAR agonists as described herein, (v) inhibitors of cholesterol absorption, such as beta-sitosterol and ezetimibe, (vi) acyl CoA cholesterol acyltransferase inhibitors, such as avasimibe, and (vii) anti-oxidants, such as probucol; (b) ileal bile acid transporter inhibitors; (c) HDL raising compounds such as CETP inhibitors or ABC1 regulators (d) lipoxygenase inhibitors; (e) ACAT inhibitors such as avasimibe; (f) fibric acid derivatives i.e. fibrates (e.g. bezafibrate, fenofibrate, gemfibrozil, clofibrate, ciprofibrate, clinofibrate or the like); (g) MTP inhibitors; (h) squalene synthetase inhibitors and squalene epoxidase inhibitors; (i) upregulators of LDL receptor activity; (j) serum cholesterol lowering agents; (k) thyroid hormone receptor agonists (sodium liothyronine, sodium levothyroxine or the like); (1) carnitine palmitoyltransferase inhibitors (etomoxir or the like); (m) probcol and microsomal triglyceride transfer protein inhibitors.
Examples of suitable anti-obesity compounds for use in combination with the compounds of the present invention include but are not limited to (a) fenfluramine, dexfenfluramine, phenteimine, tetrahydrolipostatin, and the like; (b) neuropeptide Yi or Y5 antagonists; (c) CB-1 receptor inverse agonists and antagonists; (d) β3 adrenergic receptor agonists; (e) melanocortin receptor agonists, in particular melanocortin-4 receptor agonists; (f) ghrelin antagonists; (g) melanin-concentrating hormone (MCH) receptor antagonists; (h) lipase inhibitors like orlistat; (i) serotonin (and dopamine) reuptake inhibitors like sibutramine, topiramate or axokine; (j) thyroid hormone receptor beta drugs; (k) anorectic agents like dexamphetamine, phentermine or mazindol; (1) Leptin analogs.
Examples of suitable appetite suppressants for use in combination with the compounds of the present invention include but are not limited to (a) monoamine reuptake inhibitors; (b) dopamine agonists; (c) leptin analogues; (d) ot-melanocyte stimulating hormone; (e) enterostatin agonists; (f) CCK-A agonists; (g) corticotropin releasing hormone; (h) somatostatin; (i) brain-derived neurotrophic factor; (j) orexin receptor agonists.
Examples of suitable anti-hypertensive agents for use in combination with the compounds of the present invention include but are not limited to (a) vasopeptidase inhibitors like Neutral endopeptidase (neprilysin) inhibitors and/or ACE (angiotensin- converting enzyme) inhibitors or dual NEP/ACE inhibitors (enalapril, lisinopril, captopril, quinapril, trandolapril, fosinpril, benazepril, ramipril, enalaprilat, moexipril or perindopril and the like) and/or PKC inhibitors; (b) beta blockers (like metoprolol, propranolol,
atenolol, carvedilol or sotalol) and calcium channel blockers (like amlodipine, diltiazem, nifedipine, verapamil or nicardipine ); (c) Angiotensin-II receptor blockers (like losartan, candesartan, irbesartan, valsartan, telmisartan or eprosartan); (d) Renin inhibitors e.g., aliskiren; (e) alpha blockers like terazosin, doxazosin or prasozin; (f) diuretics such as hydrochlorothiazide, torasemide, furosemide, spironolactone or indapamide; (g) thrombocyte aggregation inhibitors; (h) endothelin-converting enzyme inhibitors and endothelin receptor antagonists; (i) vasodilating antihypertensive agents e.g. indapamide, todralazine, hydralazine, budralazine or the like; (j) centrally acting antihypertensive agents e.g. reserpine; (k) a2-adrenoreceptor agonists e.g. clonidine, methyldopa, moxonidine or the like.
Examples of suitable anti-inflammatory agents for use in combination with the compounds of the present invention include but are not limited to aspirin, non-steroidal anti-inflammatory drugs, glucocorticoids, azulfidine, and selective cyclooxygenase-2 inhibitors.
Examples of suitable anti-platelet agents for use in combination with the compounds of the present invention include but are not limited to abciximab, ticlopidine, eptifibatide, dipyridamole, aspirin, anagrelide, tirofiban or clopidogrel.
Examples of suitable agents to be used in combination with the compounds of the present invention, for the treatment or prevention of hyperuricemia or gout include but are not limited to (a) uric acid synthesis inhibitors e.g. allopurinol, oxypurinol or the like; (b) uricosuric agents e.g. benzbromarone, probenecid or the like; (c) urinary alkaiiizers e.g. sodium hydrogen carbonate, potassium citrate or the like.
EXAMPLES
The invention is explained in detail in the following examples which are given solely for the purpose of illustration only and therefore should not be construed to limit the scope of the invention. Those skilled in the art will readily understand that known variations of the conditions and processes of the following preparative procedures can be used to prepare the compounds of the present invention. All of the starting materials are either commercially available or can be prepared by procedures that would be well known to one of ordinary skill in organic chemistry.
All melting points are uncorrected and expressed in °C. All solvents used in reactions were freshly distilled. Solvents were dried prior to use wherever necessary by
standard methods (Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, Pergamon Press: Oxford, 1988). Mass spectra (MS) were obtained by electron spray ionization (ESI) eV using Applied biosystem 4000 Q TRAP. Ή NMR were recorded on Bruker 400 MHz Avance II NMR spectrometer in CDC13 (until and unless specified). Chemical shifts are reported as δ values in parts per million (ppm), relative to TMS as internal standard. All coupling constant (J) values are given in Hz.
Abbreviations
The following abbreviations are employed in the examples and elsewhere herein:

MeOH methanol
mg milligram
MHz mega hertz
min minutes
mL milliliter
mmol millimoles
NaOH sodium hydroxide
Na2S04 sodium sulphate
NaHC03 sodium bicarbonate
"BuLi «-Butyl lithium
NH4C1 ammonium chloride
NMR nuclear magnetic resonance
NEt3 triethylamine
NaH sodium hydride
Pd/C palladium on carbon
Pet. ether petroleum ether pH potential hydrogen
KH potassium hydride ppm parts per million r. t. room temperature s singlet
t triplet
TBS ert-butyldimethylsilane
¾uLi ieri-Butyl lithium
THF tetrahydrofuran
TLC thin layer chromatography
Microgram
EXAMPLE I: Preparation of N-(2-(4-(2-chloro-5-((2S,3R,4R,5S,6S)-3,4,5-trihydroxy- 6-methoxytetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyI)methanesulfonamide

OH
Step 1: Preparation of ((3fl5',51S',6^,6a1S -6-((/ert-butyldimethylsilyl)oxy)-2,2-dimethyl tetrahydrofuro[2,3-< ][l,3]dioxol-5-yl)(3-(4-((tert-butyldirnethylsilyl)oxy)benzyl)-4- chlorophenyl)methanol
A solution of (4-(5-bromo-2-chlorobenzyl)phenoxy)(ieri-butyl)dimethylsilane (14.2 g, 34.57 mmol, prepared following the procedure given in US20070049537) in dry THF (90 mL) was added to LiCl (1.93 g, 46.1 mmol). The resulting solution was cooled to -78 °C and "BuLi (30.7 mL, 46.05 mmol, 1.5 M solution in hexane) was added drop wise while stirring and stirring was continued for further 30 min. A solution of (3aS,5R,6R,6aS)-6-((tert-buty\di Gt y\si\y\) oxy)-2,2-dimethyltetrahydrofuro[2,3- d][l,3]dioxole-5-carbaldehyde (7.0g, 23.05 mmol, prepared according to the procedure given in NUCLEOSIDE, NUCLOETIDES & NUCLEIC ACIDS, 20(4-7), 649-652 (2001)) in dry THF (13 mL) was added drop wise to the reaction mixture and stirred at the same temperature for 2 h. Reaction temperature was raised slowly to 0 °C and then stirred at r.t. for 16 h. After completion of reaction, as confirmed by TLC, reaction mixture was quenched by the addition of saturated NH4C1 solution and extracted with ethyl acetate (3 x 300 mL). The combined organic layers were dried over Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :49 acetone: Pet. Ether) to afford the title compound (3.66 g, 25%)
ESIMS (m/z): 657.0 (M+23)
Step 2: Preparation of (35,4i?,55,65)-6-(3-(4-acetoxybenzyl)-4-chlorophenyl)tetrahydro- 2H-pyran-2,3,4,5-tetrayl tetraacetate
A solution of ((3a5,,55,6^,6a5)-6-((teri-butyldimethylsilyl)oxy)-2,2-dimethyl tetrahydrofuro [2,3-J][l ,3] dioxol-5-yl)(3-(4-((ieri-butyldimethylsilyl)oxy) benzyl)-4- chlorophenyl) methanol (3.66 g, 5.76 mmol) in 3:2 acetic acid and water (30 mL) was refluxed at 1 10 °C for 22 h. After completion of reaction, as confirmed by TLC, reaction mixture was concentrated in vacuo. Toluene (3 10 mL) was added and distilled and the residue obtained was dissolved in pyridine (15 mL). The resulting mixture was treated
with acetic anhydride (4.0 mL, 42.62 mmol) at r.t. for 16 h. After completion of reaction, as confirmed by TLC, water was added and stirred for 1 h. The reaction mixture was diluted with ethylacetate ( 00 mL) and washed with saturated NaHC03 solution (50 mL) and brine (50 mL). The organic layers were dried over Na2S04 and the volatiles were removed in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :4 acetone: Pet.ether) to afford the title compound (2.03g, 70%)
ESI S (m/z): 598.3 (M+23)
Step 3: Preparation of (25',35',4JR,55,65)-2-bromo-6-(4-chloro-3-(4-hydroxybenzyl) phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To (35',4i?,55,65)-6-(3-(4-acetoxybenzyl)-4-chlorophenyl)tetrahydro-2H-pyran-
2,3,4,5-tetrayl tetraacetate (2.03 g, 4.03 mmol) 33% solution of HBr in AcOH (6 mL) was added at r.t. and stirred for 1 h. The reaction mixture was diluted with DC (20 mL) and stirred for 30 min. Water was added, to the resulting mixture and stirred for another 1 h, diluted it with DCM (50 mL), washed with water (50 mL), saturated NaHC03 solution (50 mL) and brine (50 mL). The organic layer was dried over Na2S04 and the volatiles were evaporated in vacuo to afford the title compound (2.02 g, 90%)
ESIMS (m/z): 554.9 (M+l)
Step 4: Preparation of (25,31S,4JR,55,65 -2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- methoxytetrahydro-2H-pyran-3 ,4, 5 -triyl triacetate
To a solution of (2S,3S,4i?,5S,6S)-2-bromo-6-(4-chloro-3-(4- hydroxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (2.02g, 3.62 mmol) in methanol (15 mL) was added ZnO (294.5 mg, 3.62 mmol) at 60 °C. The reaction mixture was stirred at 60 °C for 1 h. After completion of reaction, as confirmed by TLC, reaction mixture was passed through sintered funnel to remove the solids. The filerate was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :4 acetone: Pet. ether) to afford title compound (1.1 g, 65%)
ESIMS (m/z): 506.0 (M-l)
Step 5: Preparation of (2S, S, 4R,5S,6S)-2- (3-(4-(2-((ter/-butoxycarbonyl) amino) ethoxy) benzyl)-4-chlorophenyl)-6-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate To a solution of (25,35,4i?,55,65)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate (1.1 g, 2.35 mmol) in dry DMF (10 mL), ieri-butyl (2-bromoethyl)carbamate (1 .05 g, 4.7 mmol) and cesium carbonate (2.29 g, 7.05 mmol) were added. The reaction mixture was stirred at r.t. for 16 h. After completion of reaction, as confirmed by TLC, water (100 mL) was added to the reaction
mixture and extracted with ethylacetate (3 x 200 mL). The combined organic layers were dried over anhydrous Na2S04 and volatiles were evaporarted in vacuo. The residue obtained was purified by column chromatography (silica gel, 3:7 ethylacetate: Pet. ether) to afford the title compound (1.19 g, 85%)
ESIMS (m/z): 650.8 (M+l)
Step 6: Preparation of (2-5',35,4 ?,55,,6lS)-2-(3-(4-(2-((fert-butoxycarbonyl)amino) ethoxy)benzyl)-4-chlorophenyl) -6-methoxytetrahydro-2H-pyran-3 ,4,5-triyl triacetate
To a solution of (25,,35,4/?,55,65')-2-(3-(4-(2-((teri-butoxycarbonyl)amino)ethoxy) benzyl)-4-chlorophenyl)-6-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate (1.19 g, 1.84 mmol) in dry DC (10 mL) was added, trifluoroacetic acid (5.52 mL) at 0 °C and mixture stirred at r. t. for 2 h. After completion of reaction, as confirmed by TLC, the reaction mixture was evaporated in vacuo to afford the title compound (0.97 g, 80%).
ESIMS (m/z): 550.7 (M+l , for free amine)
Step 7: Preparation of (21S',35,4i?,55,65)-2-(4-chloro-3-(4-(2-(methylsulfonamido) ethoxy)benzyl)phenyl)-6-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (2S,3S,4tf,5S,6S)-2-(3-(4-(2-((tert-butoxycarbonyl) amino)ethoxy)benzyl)-4-chlorophenyl)-6-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate (170 mg, 0.26 mmol) in dry DCM (6 mL), triethylamine (0.1 1 mL, 0.77 mmol) and methanesulfonylchloride (0.03 mL, 0.38 mmol) were added at 0 °C and stirred for 4 h. After completion of reaction, as confirmed by TLC, water (20 mL) was added to the reaction mixture and extracted with DCM (3 x 30 mL). The combined organic layers were dried over Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 3:7Acetone: Pet. ether) to afford the title compound (130 mg, 80%)
ESIMS (m/z): 626.7 (M-l )
Step 8: Preparation of N-(2-(4-(2-chloro-5-((25,3i?,4 ?,55,65)-3,4,5-trihydroxy-6- methoxytetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)methanesulfonamide
To a solution of (25,3iS',47?,55,65 -2-(4-chloro-3-(4-(2-(methylsulfonamido)ethoxy) benzyl)phenyl)-6-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate (130 mg, 0.21 mmol) in MeOH:THF:Water (3 :2: 1 , 2.0 mL) lithium hydroxide monohydrate (17.4 mg, 0.41 mmol) was added. The reaction mixture was stirred at room temperature for 2 h. After completion of reaction, as confirmed by TLC, the reaction mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 : 15 MeOH: DCM) to afford title compound (60 mg, 59%) as off-white solid.
Ή NMR (400 MHz, CD3OD, δ): 2.97 (s, 3H), 3.29-3.25 (m, 1H), 3.45-3.41 (m, 3H), 3.46 (s, 3H), 4.07-4.02 (m, 4H), 4.1 1 (d, J = 9.5 Hz, 1H), 4.29 (d, J =7.8 Hz, 1H), 6.85 (d, J = 8.6 Hz, 2H), 7.12 (d, J= 8.6 Hz, 2H), 7.27-7.23 (m, 2H), 7.35 (d, J= 8.2 Hz, 1H)
ESIMS (m/z): 524.2 (M+23)
EXAMPLE II: Preparation of N-(2-(4-(2-chloro-5-((2S,3^54R,5S,6S)-3,4,5- trihydroxy-6-methoxytetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)sulfamide

Step 1: Preparation of (2S,3S,4i?,5S,6S)-2-(4-chloro-3-(4-(2-
(sulfamoylamino)ethoxy)benzyl) phenyl)-6-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (25,,3,5,,4^,5iS,,65)-2-(3-(4-(2-((teri-butoxycarbonyl)amino) ethoxy)benzyl)-4-chlorophenyl)-6-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate (200 mg, 0.30 mmol) in 1,4 dioxane (6 mL), triethylamine (0.13.mL, 0.90 mmol) and sulfamide (43.4 mg, 0.90 mmol) were added. The reaction mixture was reflux ed at 100 °C for 48 h. Volatiles were evaporated in vacuo, the residue obtained was dissolved in ethylacetate (150 mL) and washed with water (50 mL). The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated. The residue obtained was purified by column chromatography (silica gel, 3:97 MeOH: DCM) to afford the title compound.(90 mg, 47%) as off-white solid
ESIMS (m/z): 651.8 (M+23)
Step 2: Preparation of N-(2-(4-(2-chloro-5-((25,37?,4 ?,55,65)-3,4,5-trihydroxy-6- methoxytetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)sulfamide
The title compound (90 mg, 67%) was synthesized following the procedure reported for the synthesis of N-(2-(4-(2-chloro-5-((25,3i?,4JR,5S,65)-3,4,5-trihydroxy-6- methoxytetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)methanesulfonamide starting with (2S,3S, 4i?,55,6,S)-2-(4-chloro-3-(4-(2-(sulfamoylamino)ethoxy)benzyl) phenyl)-6- methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate (75 mg, 0.12 mmol).
Ή NMR (400 MHz, CD3OD, δ): 3.26-3.28 (m, 2H), 3.37-3.39 (m, 2H), 3.41-3.43 (m, 1H), 3.47 (s, 3H), 4.02 (d, J = 6.4 Hz, 2H), 4.06 (d, J = 5.8 Hz, 2H), 4.11 (d, J = 9.6 Hz, 1H), 4.29 (d, J = 7.7 Hz, 1H), 6.84 (dd, J = 6.7 and 4.7 Hz, 2H), 7.23 (d, J = 2.1 Hz, 2H), 7.25-7.27 (m, 2H), 7.34 (d, J= 8.1 Hz, 1H).
ESIMS (m/z): 525.9 (M+23)
EXAMPLE III: Preparation of (2S,3R,4R,5S,6R)-2-(3-(4-(2-(suIfamoyIamino) ethoxy) benzyl) -4- pyran-3,4,5-triol

Step 1: Preparation of (25,3i?,45,55,6/?)-2-(3-(4-((½ri-butyldimethylsilyl)oxy)benzyl)-4- chlorophenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
To a solution of (4-(5-bromo-2-chlorobenzyl)phenoxy)(tert-butyl)dimethylsilane (67.0 g, 0.163 mol, prepared by following the procedure given in US20070049537) in dry THF (1.0 L), 'BuLi (228 mL, 0.342 mol, 1.6 M solution in pentane) was added at -78 °C. The reaction mixture was stirred at the same temperature for 30 min. A solution of (3i?,45',5i?,6i?)-3,4,5-tris((trimethylsilyl)oxy)-6-(((trimethylsilyl)oxy)methyl)tetrahydro- 2H-pyran-2-one (77.8 g, 0.167 mol, prepared following the procedure given in US20070049537) in dry THF (272 mL) was introduced into it at the same temperature and stirred for another 4 h. A solution of methanesulfonic acid (18.35 mL, 0.283 mol) in methanol (586 mL) was added and the temperature was raised to r.t. gradually and stirred for 16 h. The reaction was quenched by the addition of saturated aqueous solution of NaHC03 (500 mL) and volatiles were evaporated in vacuo. The residue obtained was dissolved in ethylacetate (1L), washed with water (500 mL) and brine (500 mL) successively. The organic layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide title compound (47.37 g, 55.4 %) as off-white solid. ESIMS (m/z): 524.2 (M-l)
Step 2: Preparation of (2S,3/?,45,5R,6R)-6-(acetoxymethyl)-2-(3-(4-((ieri- butyldimethylsilyl) oxy)benzyl)-4-chlorophenyl)-2-methoxytetrahydro-2H-pyran-3,4,5- triyl triacetate
To a solution of (25,3i?,45,55,6JR)-2-(3-(4-((tert-butyldimethylsilyl)oxy)benzyl)-4- chlorophenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (47.0 g, 0.090 mol) in dry THF (286 mL), diisopropylethylamine (114.28 mL, 0.660 mol) and 4- dimefhylaminopyridine (3.94 g, 0.032 mol) were added. Acetic anhydride (55 mL, 0.582 mol) was added to the resulting solution at 0 °C. The reaction mixture was stirred at r.t. for 2 h. Ethylacetate (1L) was added to the reaction mixture, washed it with 2% HC1 solution (2 x 100 mL), water (500 mL) and brine (500 mL) successively. The organic layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide title compound (53.9 g, 86.93 %) as off-white solid.
ESIMS (m/z): 715.0 (M+23)
Step 3: Preparation of (2i?,3JR,4i?,55,65)-2-(Acetoxymethyl)-6-(4-chloro-3-(4- hydroxybenzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (25,,3 ?,4i,,5^,6^)-6-(acetoxymethyl)-2-(3-(4-((teri- butyldimethylsilyl)oxy)benzyl)-4-chlorophenyl)-2-methoxytetrahydro-2H-pyran-3,4,5- triyl triacetate (44.0 g, 0.064 mol) in acetonitrile (200 mL), water (1.15 mL, 0.064 mol), triethylsilane (32.45 mL, 0.203 mol) and borontrifluoroetherate (15.94 mL, 0.127 mol) were added at 10 °C. The resulting mixture was stirred at r.t. for 16 h. Additional amounts of triethylsilane (3.25 mL, 0.020 mol) and borontrifluoroetherate (1.59 mL, 0.013 mol) were added at 10 °C and heated at 30 °C for 6h. Ethylacetate (1L) was added to the reaction mixture, washed it with saturated NaHC03 solution (2 x 500 mL), water (500 mL) and brine (500 mL) successively. The organic layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 Ethylacetate: Pet.ether)-to provide title compound (30 g, 86.27 %) as off-white solid.
ESIMS (m/z): 548.2 (M-l)
Step 4: Preparation of ierr-butyl (2-bromoethyl)carbamate
To a stirred suspension of 2-bromoethanamine hydrobromide (15 g, 0.073 mol) in dry DCM (293 mL), triethylamine (30.61 mL) was added at 0 °C. The resulting mixture was treated with di fcr -butyl dicarbonate (25.23 mL, 0.01 lmol), gradually raised the temperature to r.t. and stirred at that temperature for 16 h. The reaction mixture was diluted with DCM (200 mL) and washed with water (2 x 200 mL) and brine (200 mL) successively. Organic layer was separated, dried over anhydrous Na2S04 and the volatiles
were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 ethylacetate: Pet. ether) to provide title compound (7.5 g, 46 %) as an oil. ESIMS (m/z): 224.6 (M+lof 79Br), 226.6 (M+lof 8lBr)
Step 5: Preparation of (2/?,3/?,47?,55,65)-2-(acetoxymethyl)-6-(3-(4-(2-((tert- butoxycarbonyl) amino)ethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3 ,4,5-triyl triacetate
To a solution of (2 ?,3^,4J?,55,65)-2-(Acetoxymethyl)-6-(4-chloro-3-(4 hydroxy benzyl) phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (5 g, 0.009 mol) and 2- bromoethahamine hydrobromide (4.08 g, 0.018 mol) in dry DMF (18 mL), cesium carbonate (4.45 g, 0.014 mol) was added at r.t. The reaction mixture was stirred at r.t. and progress of the reaction was monitored by TLC. On completion, the resulting mixture was diluted with ethylacetate (200 mL) and washed with water (2 x 200 mL) and brine (200 mL) successively. Organic layer was separated, dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :4 acetone: Pet. ether) to provide title compound (5.7 g, 90 %) as off-white solid.
ESIMS (m/z): 731.0 (M+39), 714.9 (M+23), 692.9 (M+l)
Step 6: Preparation of (2 ?,3i?,4/?,55,,65)-2-(acetoxymethyl)-6-(3-(4-(2- aminoethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3 ,4,5-triyl triacetate 2,2,2- trifiuoroacetate
To a solution of (2JR,3^,4i?,55,61S)-2-(acetoxymethyl)-6-(3-(4-(2-((fer/- butoxycarbonyl) amino)ethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (5.7 g, 0.008 mol) in dry DCM (16.5 mL), trifluoroaceticacid (24.75 mL) was added at 0 °C. The resulting mixture was stirred while gradually raising the temperature to r.t. for 1 h. Volatiles were evaporated in vacuo and the residue obtained was washed with diethylether (2 x 20 mL) to provide title compound (5.7 g, 97.93 %) as off-white solid. ESIMS (m/z): 592.6 (M+l, for free amine)
Step 7: Preparation of (2^,37?,4J/?,5^,6 ,)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2 (sulfamoylamino)ethoxy)benzyl) phenyl)tetrahydro-2H-pyran-3 ,4,5-triyl triacetate
To ' a solution of ((2#,3^4/?,5S,6S 2-(acetoxymethyl)-6-(3-(4-(2- aminoethoxy)benzyl)-4-chlorophenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate 2,2,2- trifluoroacetate (1 g, 0.0014 mol) in dry dioxane (7.08 mL), triethylamine (0.5 mL, 0.004 mol) was added at 0 °C and stirred while gradually raising the temperature to r.t. Sulfamide (0.204 g, 0.002 mol) was added and the resulting mixture was refluxed for 4 h.
Dioxane was evaporated in vacuo and the residue obtained was diluted with ethylacetate (50 mL). Organic layer was washed with water (20 mL) and brine (20 mL) successively. Ethylacetate layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 acetone: Pet. ether) to provide the title compound (0.525 g, 55 %) as white solid.
ESIMS (m/z): 703.0 (M+23), 671.8 (M+l)
Step 8: Preparation of (25,3i?,4i?,55,6i?)-2-(3-(4-(2-(sulfamoylamino)ethoxy)benzyl)-4- chlorophenyl)-tetrahydro-6-(hydroxymethyl)-2H-pyran-3 ,4,5-triol
To a solution of (2Λ,3Λ,4Λ,55,65 -2-(αϋείοχ^ε 1)-6-(4-ϋΜοΓθ-3-(4-(2- (sulfamoylamino) ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (525 mg, 0.783 mmol) in THF (2.6 mL), methanol (3.9 mL) and water (1.3 mL) was added lithium hydroxide monohydrate (50 mg, 1.17 mmol) at 0 °C. The reaction was stirred at r.t. for 1 h. Volatiles were evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :4 MeOH: DCM) to provide the title compound (350 mg, 89 %) as white solid.
Ή NMR (400 MHz, CD3OD, 5): 3.26 (d, J= 8.9 Hz, 1H), 3.46-3.33 (m, 5H), 3.67 (dd, J = 11.9 and 5.2 Hz, 1H), 3.85 (dd, J = 13.1 and 1.6 Hz, 1H), 3.98-4.09 (m, 5H), 6.83-6.86 (m, 2H), 7.10 (d, J = 8.2 Hz, 2H), 7.26 (dd, J = 8.1 and 2.0 Hz, 1H), 7.30 (d, J = 1.9 Hz, 1H), 7.33 (d, J= 8.2 Hz, 1H).
ESIMS (m/z): 525.2 (M+23)
EXAMPLE IV: Preparation of (2S,3R,4R,55',6R)-2-(3-(4-(3-(sulfamoyIamino) propoxy)benzyI)-4-chlorophenyl)-tetrahydro-6-(hydroxymethyl)-2H-pyran-3,4,5-triol

Step 1: Preparation of teri-butyl (3-hydroxypropyl)carbamate
To a solution of 3-amino propan-l -ol (6 g, 0.080 mol) in dry DCM (390 mL), triethyl amine (22.3 mL, 0.160 mol) was added at 0 °C followed by the addition of di tert- butyl dicarbonate (27.5 mL, 0.120 mol). The resulting mixture was stirred at r.t. for 16 h and the reaction was monitored by TLC. On completion, reaction mixture was diluted with DCM (200 mL), washed with water (200 mL) and brine (200 mL) successively. The
organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide title compound (9.9 g, 71 %) as an oil.
ESIMS (m/z): 176.6 (M+l)
Step 2: Preparation of 3-((teri-butoxycarbonyl)amino)propyl methanesulfonate
To a solution of tert-butyl (3-hydroxypropyl)carbamate (4 g, 0.023 mol) in dry DCM (1 14 mL), triethyl amine was added (6.4 mL, 0.046 mol) at 0 °C followed by the addition of methanesulfonyl chloride (2.7 mL, 0.034 mol). The resulting mixture was stirred at r.t. for 2 h and the reaction was monitored by TLC. On completion, reaction mixture was diluted with DCM (100 mL), washed with water (50 mL) and brine (50 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo to afford the title compound.
ESIMS (m/z): 276.6 (M + 23), 254.8 (M + l)
Step 3: Preparation of (2i¾,3^,4^,5S,6S)-2-(acetox methyl)-6-(3-(4-(3-((tert- butoxycarbonyl) amino)propoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
The title compound (2.0 g, 55.5 %) was synthesized following the procedure reported for the synthesis of (2^,3 ?,4 ?,5-S,65)-2-(acetoxymethyl)-6-(3-(4-(2-((tert- butoxycarbonyl)amino) ethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate starting with (2^,3^,47?,55,61S)-2-(acetoxymethyl)-6-(4-chloro-3-(4- hydroxybenzyl) phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (2.8 g, 0.005 mol) and 3- ((tert-butoxycarbonyl)amino)propyl methanesulfonate (1.94 g, 0.008 mol).
ESIMS (m/z): 701.9 (M-l)
Step 4: Preparation of (2 ?,3i?,4 ?,55,65)-2-(acetoxymethyl)-6-(3-(4-(3- aminopropoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2,2,2- trifluoroacetate
The title compound (1.80 g, 98.4 %) was synthesized following the procedure reported for the synthesis of (2i ^,4i^5iS 65)-2-(acetoxymethyl)-6-(3-(4-(2- aminoethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2,2,2- trifluoroacetate starting with ((2i?,3j!?,4i?,51S,,66,)-2-(acetoxymethyl)-6-(3-(4-(3-((tert- butoxycarbonyl)amino)propoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (1.8 g, 0.003 mol).
ESIMS (m/z): 606.2 (M+ 1 , for free amine)
Step 5: Preparation of (2R AR,5S,6S)-2-(acetoxy ethy\)-6-(4-ch\oro-3-(4-Q-
(sulfamoylamino) propoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
The title compound (140 mg, 49.12 %) was synthesized following the procedure reported for the synthesis of (2i?,3JR,4i?,55',65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2 (sulfamoylamino) ethoxy)benzyl) phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate starting with (2^,3 ?,4^,55,65')-2-(acetoxymethyl)-6-(3-(4-(3-aminopropoxy)benzyl)-4- chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2,2,2-trifluoroacetate (300 mg,
0.417 mmol).
ESIMS (m/z): 685.8(M+1)
Step 6: Preparation of (25',3/?,4i?,55,6 ?)-2-(3-(4-(3-(sulfamoylamino)propoxy)benzyl)-4- chlorophenyl)-tetrahydro-6-(hydroxymethyl)-2H-pyran-3,4,5-triol
The title compound (70 mg, 66.66 %) was synthesized following the procedure reported for the synthesis of (25,3Λ,4Λ,55,.6Λ)-2-(3-(4-(2-
(sulfamoylamino)ethoxy)benzyl)-4-chlorophenyl)-tetrahydro-6-(hydroxymethyl)-2H- pyran-3,4,5-triol starting with (2 ?,3i?,4i?,55',65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(3-
(sulfamoylamino)propoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (140 mg, 0.204 mmol).
Ή NMR (400 MHz, CD3OD, δ): 1.97-2.01 (m, 2H), 3.20 (t, J = 6.9 Hz, 2H), 3.26 (d, J = 8.9 Hz, 1H), 3.36-3.43 (m, 3H), 3.67 (dd, J = 12.0 and 5.3 Hz, 1H), 3.85 (dd, J= 12.9 and 1.7 Hz, 1H), 3.96-4.04 (m, 4H), 4.07 (d, J = 9.4 Hz, 1H), 6.80-6.82 (m, 2H), 7.08 (d, J = 8.7 Hz, 2H), 7.26 (dd, J - 8.2 and 2.1 Hz, 1H), 7.30 (d, J = 1.9 Hz, 1 H), 7.33 (d, J = 8.2 Hz, 1H).
ESIMS (m/z): 516.1 (M-l) . EXAMPLE V: Preparation of N-(3-(4-(2-chloro-5-((25,3R,4R,5S,6R)-3,4,5- trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)propyl) cyclohexanesulfonamide

The title compound (100 mg, 57 %) was synthesized following the procedure reported for the synthesis of (2i?,3i?,4ii,55,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- (methylsulfonamido) ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate starting with (2 ?,3i?,4i?,55',65 -2-(acetoxymethyl)-6-(3-(4-(3-aminopropoxy)benzyl)-4- chIorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2,2,2-trifluoroacetate (150 mg, 0.204 mmol) and cyclohexanesulfonyl chloride (0.051 mL, 0.35 mmol) in place of methane sulfonyl chloride.
ESIMS (m/z): 752.9 (M+l)
Step 2: Preparation of N-(3-(4-(2-chloro-5-((25,3 ?,4i?,55,6i?)-3,4,5-trihydroxy-6-
(hydroxymethyl)tetrahydro-2H-pyran-2- yl)benzyl)phenoxy)propyl)cyclohexanesulfonamide
The title compound (70 mg, 90 %) was synthesized following the procedure reported for the synthesis of (2R,3R,4R,55,6S)-2-(3-(4-(2- (sulfamoylamino)ethoxy)benzyl)-4-chlorophenyl)-tetrahydro-6-(hydroxymethyl)-2H- pyran-3,4,5-triol starting with (2i?,3 ?, i?,55',(55)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(3- (cyclohexanesulfonamido)propoxy)benzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (100 mg, 0.133mmol).
1H NMR (400 MHz, CD3OD, δ): 1.1 1-1.28 (m, 4H), 1.37-1.46 (m, 2H), 1.79 (d, J = 12.8 Hz, 2H), 1.92-1.98 (m, 2H), 2.08 (d, J = 13.2 Hz, 2H), 2.85-2.91 (m, 1H), 3.23 (t, J= 6.7 Hz, 3H), 3.37 (d, J= 7.4 Hz, 2H), 3.41-3.47 (m, 1H), 3.67 (dd, J= 11.8 and 5.00 Hz, 1H), 3.86 (d, J= 1 1.7 Hz, 1H), 4.00-4.03 (m, 4H), 4.07 (d, J = 9.5 Hz, 1H), 6.81 (d, J - 8.6 Hz, 2H), 7.05 (d, J = 8.6 Hz, 2H), 7.26 (dd, J= 8.2 and 2.00 Hz, 1 H), 7.31-7.34 (m, 2H).
ESI-MS (m/z): 584.7 (M+l)
EXAMPLE VI: Preparation of 7V-(2-(4-(2-chloro-5-((2S,3R,4R,5S,6R)-3,4,5- trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2- l)benz l) heno )ethyl)- > !( ,- dimethylphosphinic amide

Step 1: Preparation of (2/?,3Λ,4Λ,55,65)-2-(3θ6ίοχ^ε 1)-6-(4-ε1ι1οΓθ-3-(4-(2-
((dimethylphosphoryl)amino)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (2i?,3i?,4^,56',6 )-2-(acetoxymethyI)-6-(3-(4-(2- aminoethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2,2,2- trifluoroacetate (280 mg, 0.473 mmol) in dry DCM (5 mL) was added triethylamine (0.197 mL, 1.419 mmol) at 0 °C, followed by dimethyl phosphinic chloride (79.8mg, 0.71 mmol). The reaction mixture was stirred at same temperature for 4 h. After completion of reaction, as confirmed by TLC, the reaction mixture was diluted with DCM (50 mL) and given water washings (2 x 10 mL). The organic layer was dried over anhydrous Na2S04. Volatiles were evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 2.5:97.5 MeOH: DCM) to afford title compound as a white solid (180 mg¾ 57.01%).
ESI-MS (m/z): 668.9 (M+l)
Step 2: Preparation of JV-(2-(4-(2-chloro-5-((2,S,3i?,4JR,55,6JR)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)- ,JP- dimethylphosphinic amide
To a solution of (2R,3R R,5S,6S)-2-(acetoxymethy\)-6-(4-ch}oro-3-(4-(2- ((dimethylphosphoryl)amino)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (170 mg, 0.254 mmol) in MeOH: THF: Water (3:2: 1, 2.5 mL), lithium hydroxide monohydrate (10.69 mg, 0.254 mmol) was added at 0 °C. The reaction mixture was stirred at r.t. for 1 h. After completion of reaction, as confirmed by TLC, the reaction mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :10 MeOH: DCM) to afford title compound (92mg, 72.5%) as off-white solid.
Ή NMR (400 MHz, CD3OD, δ): 1.48 (s, 3H), 1.52 (s, 3H), 3.24-3.29 (m, 3H), 3.37-3.38 (m, 2H), 3.40-3.44 (m, 1H), 3.65-3.69 (m, 1 H), 3.84-3.87 (m, 1H), 3.97 (t, J = 5.4 Hz, 2H), 4.01 -4.08 (m, 3H), 6.82-6.84 (m, 2H), 7.09-7.1 1 (m, 2H), 7.26 (dd, J = 8.2 and 2.1 Hz, 1H), 7.31-7.34 (m, 2H).
ESI-MS (m/z): 500.6 (M+l)
EXAMPLE VII: Preparation of dimethyl (3-(4-(2-chloro-5-((25,3R,4R,5S,6R)-3,4,5- trihydroxy-6-(hydroxymethyI)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)propyl) phosphoramidate

Step 1: Preparation of dimethyl (3-hydroxypropyl) phosphoramidate
To a solution of 3-amino propan-l -ol (500 mg, 6.65 mmol) in dry DCM (33 mL), triethyl amine (1.85 mL, 13.31 mmol) was added at 0 °C followed by the addition of dimethyl phosphorochloridate (0.72 mL, 6.65 mmol). The resulting mixture was stirred at r.t. for 2 h and the reaction was monitored by TLC. On completion, volatiles were evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide title compound (340 g, 28 %) as an oil.
ESIMS (m/z): 184.7 (M+l)
Step 2: Preparation of 3-((dimethoxyphosphoryl)amino)propyl methanesulfonate
The title compound (456 mg, 100 %) was synthesized following the procedure reported for the synthesis of 3-((teri-butoxycarbonyl)amino)propyl methanesulfonate starting with dimethyl (3-hydroxypropyl)phosphoramidate (320 mg, 1.74 mmol).
ESIMS (m/z): 262.5 (M + l)
Step 3: Preparation of dimethyl (3-(4-(2-chloro-5-((25,37i,4/?,55,6 ?)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)propyl)phosphoramidate
To a solution of (25,3i?,4 ?,55,6i?)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (200 mg, 0.53 mmol, prepared according to the procedure given in WO2009026537) and 3-((dimethoxyphosphoryl)amino)propyl methanesulfonate (274 mg, 1.05 mmol) in dry DMF (1.3 mL), cesium carbonate (513 mg, 1.57 mmol) and sodium iodide (15.7 mg, 0.11 mmol) were added at r.t. The reaction mixture was stirred at the same temperature and progress of the reaction was monitored by TLC. On completion, the resulting mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide title compound (100 mg, 35 %) as off white solid.
Ή NMR (400 MHz, CD3OD, δ): 1.90-1.94 (m, 2H), 3.02-3.08 (m, 2H), 3.36-3.47 (m, 4H), 3.62 (s, 3H), 3.64 (s, 3H), 3.66-3.69 (m, 1H), 3.85 (d, J= 11.7 Hz, 1H), 3.96-4.02 (m, 4H), 4.07 (d, J = 9.4 Hz, 1H), 6,81 (d, J = 8.6 Hz, 2H), 7.08 (d, J = 8.6 Hz, 2H), 7.25 (dd, J = 8.2 and 2.1 Hz, 1H), 7.30-7.34 (m, 2H).
ESI-MS (m/z): 546.9 (M+l)
EXAMPLE VIII: Preparation of N-(4-(2-chloro-5-((2S,3R,4R,5S,6R)-3,4,5- trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenethyl)sulfamide

Step 1: Preparation of methyl 5-bromo-2-chlorobenzoate
To a solution of 5-bromo-2-chlorobenzoicacid (20.0 g, 0.085 mol) in dry DCM (100 mL) and DMF (0.1 mL), oxalylchloride (7.63 mL, 0.89 mol) was added drop wise at 0 °C. The resulting mixture was allowed to stir at r.t. for 16 h. Volatiles were evaporated and the residue obtained was dissolved in dry DCM (425 mL). A solution of methanol (424 mL) and triethylamine (35.5 mL, 0.254 mmol) was added to it at 0 °C and stirred at the same temperature for 1 h. Reaction mixture was washed with water (200 mL) and brine solution (200 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :9 ethylacetae: Pet.ether) to provide the title compound (20 g, 94.4 %) as pale yellow solid.
ESI-MS (m/z): 271.0 (M+23), 249.1 (M+l)
Step 2: Preparation of 5-bromo-2-chlorobenzaldehyde
To a solution of methyl 5-bromo-2-chlorobenzoate (6 g, 24.04 mmol) in dry DCM (120 mL), diisobutylaluminium hydride in toluene (36 mL, 36 mmol, 1M solution) was added at -78°C and allowed to stir for 30 min. The reaction was quenched by the addition of methanol (4.8 mL) and IN HCl (154 mL), diluted with DCM (150 mL). The organic layer was separated and dried over anhydrous Na2S04 and the volatiles were evaporated. The residue obtained was purified by column chromatography (silica gel, 1 :49 ethylacetate: Pet.ether) to provide the title compound (3.2 g, 60.6 %) as pale yellow solid. ESI-MS (m/z): 236.3 (M+l 8)
Step 3: Preparation of l-(2-(Allyloxy)ethyl)-4-bromobenzene
To a suspension of 60 % sodium hydride (7.95 g, 0.20 mol) in dry THF (125 mL), a solution of 2-(4-bromophenyl)ethanol (10 g, 0.05 mol) in dry THF (125 mL) was added slowly at 0 °C in 10 min. Allyl bromide (13.5 mL, 0.15 mol) was added to the resulting mixture after 10 min. The reaction mixture was stirred for 3 h while monitored by TLC. On completion, the reaction mixture was quenched by the addition of brine solution (lmL) at 0 °C. It was diluted with ethyl acetate (500 mL), washed with water (100 mL) and brine (100 mL) successively. Organic layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :9 ethyl acetate: Pet.ether) to provide title compound (12.0 g, 100 %) as an oil. ESI-MS (m/z): 279.4 (M+39)
Step 4: Preparation of 2-(4-(2-(allyloxy)ethyl)benzyl)-4-bromo-l -chlorobenzene
Step 4.1: To a solution of l-(2-(Allyloxy)ethyl)-4-bromobenzene (1 g, 4.14 mmol) in dry
THF (10 mL) (BuLi (6.07 mL, 9.12 mmol, 1.5 M in pentane) was added at -78 °C and allowed to stir for 15 min. A solution of 5-bromo-2-chlorobenzaldehyde (910 mg, 4.14 mmol) in dry THF (10 mL) was added to the resulting mixture and stirred at the same temperature for 3 h. The reaction was quenched by the addition of saturated NH4C1 solution (5 mL). It was diluted with ethylacetate (50 mL) and washed with water (20 mL) and brine solution (20 mL) successively. The organic layer was separated and dried over anhydrous Na2S04 and the volatiles were evaporated. The residue obtained was obtained was purified by column chromatography (silica gel, 1 :9 ethylacetate: Pet.ether) to provide off-white solid (580 mg, 38.6 %) which was used as such in the next step {step 4.2).
Step 4.2: To a solution of (4-(2-(Allyloxy)ethyl)phenyl)(5-bromo-2- chlorophenyl)methanol (5.1 g, 13.36 mmol) in acetonitrile (40 mL) triethylsilane (6.82 mL, 42.75 mmol) was added at 10°C followed by the addition of boranetrifluoroetherate (3.35 mL, 26.72 mmol). The resulting mixture was stirred at r.t. for 4 h. The reaction was diluted with ethylacetate (100 mL), washed with water (50 mL), saturated NaHC03 solution (50 mL) and brine solution (50 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated. The residue obtained was purified by column chromatography (silica gel, 1 :9 ethylacetae: Pet.ether) to provide the title compound (4 g, 81.79 %) as off-white solid.
ESI-MS (m/z): 365.7 (M+l)
Step 5: Preparation of ' (25,3^,45,5i?,6K)-6-(acetoxymethyl)-2-(3-(4-(2- (allyloxy)ethyl)benzyl)-4-chlorophenyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triyl triacetate
Step 5.1: To a solution of 2-(4-(2-(allyloxy)ethyl)benzyl)-4-bromo-l-chlorobenzene (300 mg, 0.818 mmol) in dry THF (5 mL), "BuLi (0.82 mL, 1.23 mmol, 1.5 M in hexane) was added at -78 °C and allowed to stir for 5 min. A solution of (3i?,45,,5i?,6i?)-3,4,5- tris((trimethylsilyl)oxy)-6-((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one (390 mg, 0.834 mmol) in dry THF (1.6 mL) was added to the resulting mixture and stirred at the same temperature for 4 h. The reaction was quenched by the addition of a solution of methanesulfonicacid (0.14 mL, 2.12 mmol) in methanol (4 mL) and stirred at r.t. for 16 h. Triethylamine (0.34 mL) was added to it and volatiles were evaporated in vacuo. The residue obtained was dissolved with ethylacetate (50 mL) and washed with water (20 mL) and brine solution (20 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated. The residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide (25,3i?,45,55,67?)-2-(3-(4-(2- (allyloxy)ethyl)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H- pyran-3,4,5-triol (110 mg, 28.2 %) as an off-white solid.
Step 5.2: To a solution of (25,3i?,45,55,6 ?)-2-(3-(4-(2-(allyloxy)ethyl)benzyl)-4- chlorophenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (3.86 g, 8.05 mmol) in dry THF (42 mL), diisopropylethylamine (10.29 mL, 59.07 mmol), 4- •dimethylaminopyridine (354 mg, 2.89 mmol) were added. Acetic anhydride (4.94 mL, 52.32 mmol) was added to the resulting solution at 0 °C. The reaction mixture was stirred at r.t. for 2 h. Ethylacetate (250 mL) was added to the reaction mixture, washed it with 5% HC1 solution (100 mL), water (100 mL) and brine (100 mL) successively. The organic layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide title compound (5.21 g, 100 %) as off-white solid.
ESI-MS (m/z): 670.0 (M+23)
Step 6: Preparation of (2i?,3i?,4i?,5S,6S)-2-(acetoxymethyl)-6-(3-(4-(2- (allyloxy)ethyl)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (25,37?,45,5^,6Λ)-6-(3ϋεΐοχ^6 1)-2-(3-(4-(2- (allyloxy)ethyl)benzyl)-4-chlorophenyl)-2-methoxytetrahydro-2H-pyi-an-3,4,5-triyl triacetate (1.0 g, 1.54 mmol) in acetonitrile (4.62 mL), water (0.027 mL, 1.54 mmol), triethylsilane (0.79 mL, 4.94 mmol) and borontrifluoroetherate (0.39 mL, 3.08 mmol)
were added at 10 °C. The reaction mixture was stirred at r.t. for 18 h. Additional amounts of triethylsilane (0.39 mL, 2.47 mmol) and borontrifluoroetherate (0.19 mL, 1.54 mmol) were added at 10 °C and stirred for 24 h. Ethylacetate (40 mL) was added to the reaction mixture, washed it with saturated NaHC03 solution (2 x 20 mL), water (20 mL) and brine (20 mL) successively. The organic layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 Ethylacetate:Pet.ether) to provide title compound (800 mg, 84.2 %) as off- white solid.
ESI-MS (m/z): 638.4 (M+23)
Step 7: Preparation of (2i?,3i?,4/?,51S,65 -2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- hydroxyethyl)benzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate
A solution of (2i?,3/?,4^,55,65)-2-(acetoxymethyl)-6-(3-(4-(2-
(allyloxy)ethyl)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (1 g,
1.62 mmol) in dioxane (10.8 mL) and water (1.08 mL) was treated with N- methylmorpholine-N-oxide (854 mg, 7.29 mmol), sodiumperiodate (1.56 g, 7.29 mmol) in water (4.4 mL) and osmiumtetroxide (3 mL, 3 mmol, 1M solution in tert-butanol) at r.t.
The resulting mixture was heated at 60 °C for 42 h. It was extracted with DCM (3 x 50 mL). The combined organic layer was washed with brine (20 mL), dried over anhydrous
Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 Ethylacetate: Pet.ether) to provide title compound
(640 mg, 68.8 %) as off-white solid.
ESI-MS (m/z): 600.2 (M+23)
Step 8: Preparation of (2 ?,3R,4R,5S,6S)-2-(acetox methyl)-6-(4-chloro-3-(4-(2- ■ ((methylsulfonyl)oxy)ethyl)benzyl)phenyl)tetrahydro-2H-pyran-3 ,4,5 -triyl triacetate
To a solution of (2i?,3 ?,4i?,55,61S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- hydroxyethyl)benzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (448 mg, 0.776 mmol) in dry DCM (3.88 mL), triethyl amine was added (0.22 mL, 1.55 mmol) at 0 °C followed by the addition of methanesulfonyl chloride (0.09 mL, 1.16 mmol). The resulting mixture was stirred at r.t. for 3 h and the reaction was monitored by TLC. On completion, reaction mixture was diluted with DCM (20 mL), washed with water (10 mL) and brine (10 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo to afford the title compound which was used as such in further steps.
ESI-MS (m/z): 677.8 (M+23)
Step 9: Preparation of (2 ?,3JR,4i?,55,6lS)-2-(acetoxymethyl)-6-(3-(4-(2-azidoethyl)benzyl)- 4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (2Λ,3Λ,4Λ,55,65)-2-( ο6ίοχ^6 1)-6-( -ΰ1ι1θΓθ-3-(4-(2- ((methylsulfonyl)oxy)ethyl)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (530 mg, 0.81 mmol) in dry DMF (4.0 mL), sodium azide (262 mg, 4.04 mmol) was added and the resulting solution was heated to 60 °C while stirring for 16 h. The reaction mixture was diluted with ethyl acetate (50 mL), washed with water (20 mL) and brine (20 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 ethylacetate: Pet.ether) to provide the title compound (390 mg, 81.3 %) as white solid.
ESI-MS (m/z): 624.9 (M+23)
Step 10: Preparation of (2i?,3i?,4 ,5S,6S)-2-(acetoxymethyl)-6-(3-(4-(2- aminoethyl)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (2i ^^R,5S,6S)-2-(acetoxymethyl)-6-(3-(4-(2- azidoethyl)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (380 mg, 0.631 mmol) in THF: water (1 : 1 , 5 mL), triphenylphosphine (182 mg, 0.69 mmol) was added. The resulting solution was stirred at r.t. for 16 h. The volatiles were evaporated and the residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide the title compound (240 mg, 66.6 %) as white solid.
ESI-MS (m/z): 576.7 (M+l)
Step 11. Preparation of (2^,3^,4^,5- ,,65,)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- (sulfamoylamino)ethyl)benzyl)phenyl) tetrahydro-2H-pyran-3 ,4,5-triyl triacetate
To a solution of ((2i?,3i?,4 ?,55,65)-2-(acetoxymethyl)-6-(3-(4-(2- aminoethyl)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (340 mg, 0.59 mmol) in dry dioxane (3.0 mL), triethylamine (0.21 mL, 1.47 mmol) was added at 0 °C and stirred while gradually raising the temperature to r.t. Sulfamide (85 mg, 0.89 mmol) was added and the resulting mixture was refluxed for 3 h. Dioxane was evaporated in vacuo and the residue obtained was diluted with ethylacetate (50 mL). Organic layer was wahed with water (20 mL) and brine (20 mL) successively. Ethylacetate layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 acetone: Pet. ether) to provide the title compound (0.17 mg, 44.73 %) as white solid.
ESI-MS (m/z): 655.9 (M+l)
Step 12: Preparation of N-(4-(2-chloro-5-((25,3i?,4i?,55,67?)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenethyl)sulfamide
To a solution of (2R,3R,4R,5S S)-2-(acetoxymet yl)-6-(4-chloro-3-(4-(2- (sulfamoylamino)ethyl)benzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (170 mg, 0.26 mmol) in THF (0.86 mL), methanol (1.3 mL) and water (0.43 mL) was added lithium hydroxide monohydrate (11 mg, 0.22 mmol) at 0 °C. The reaction was stirred at r.t. for 1 h. Volatiles were evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide the title compound (90 mg, 71.4 %) as white solid.
Ή NMR (400 MHz, CD3OD, δ): 2.81 (t, J = 7.6 Hz, 2H), 3.21 (t, J= 7.6 Hz, 2H), 3.26 (d, J = 9.0 Hz, 1H), 3.37-3.47 (m, 3H), 3.66-3.69 (m, 1H), 3.86 (d, J = 1 1.8 Hz, 1H), 4.01- 4.1 1 (m, 3H), 7.13 (s, 4H), 7.27 (dd, J = 8.2 and 2.0 Hz, 1H), 7.30 (d, J = 1.9 Hz, 1H), 7.33 (d, J= 8.2 Hz, 1H), 7.32-7.35 (m, 2H).
ESI-MS (m/z): 487.7 (M+l )
EXAMPLE IX: Preparation of N-(2-(4-(2-chloro-5-((2S,3R,4R,5S,6R)-3,4,5- trihydroxy-6-(hydroxymethyl)tetrahydro-2 7-pyran-2- yl)benzyl)phenoxy)ethyl)pyrrolidine-l-sulfonamide

Step 1: Preparation of (2 ?,3/?,4^,55,65)-2-(acetoxymethyl)-6-(3-(4-(2- aminoethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (2/.,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(3-(4-(2- aminoethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2,2,2- trifluoroacetate (780 mg, 1.1 mmol) in dry DCM (15 mL), triethylamine (0.46 mL, 3.31 mmole) was added at 0 °C. The reaction mixture was stirred at r.t. for 24 h. The reaction mixture was diluted with DCM (150 mL) and washed with water (5x) 0 mL). The organic layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo to afford the title compound (600 mg, 92%).
ESIMS (m/z): 593 (M+l)
Step 2: Preparation of (2i?,3i?,4/?,55,61S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- (pyrrolidine- 1 -sulfonamido)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3 ,4,5-triyl triacetate
To a solution of (2i ^,4^5S,6S)-2-(acetoxymethyl)-6-(3-(4-(2- aminoethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (100 mg, 0.168 mmol) in 2,6-leutidine (0.2 mL), pyrrolidine- 1-sulfonyl chloride (28.6 mg, 0.168 mmol) was added at r.t. The resulting mixture was stirred at r.t. for 16 h and the reaction was monitored by TLC. On completion, reaction mixture was diluted with ethyl acetate (20 mL), washed with 5 % HC1 (2 x 10 mL), water (10 mL) and brine (10 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 3:7 ethylacetate: Pet. ether) to provide the title compound (80 mg, 65.5 %) as white solid.
ESIMS (m/z): 747.3 (M+23)
Step 3: Preparation of N-(2-(4-(2-chloro-5-((25,3i?,4i?,55,6i?)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)pyrrolidine-l- sulfonamide
To a solution of (2R,3R,4/.,5S,6.S)-2-(acetox mei¾yl)-6-(4-cWoiO-3-(4-(2- (pyrrolidine-l-sulfonamido)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (150 mg, 0.21 mmol) in methanol (1.02 ml), THF (0.68 mL) and water (0.34 mL), lithium hydroxide monohydrate (8.6 mg, 0.21 mmol) was added at 0 °C. The resulting mixture was stirred at r.t. for 1 h. Volatiles were evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide the title compound (60 mg, 52.2 %) as white solid.
Ή NMR (400 MHz, CD3OD, δ): 1.84-1.88 (m, 4H), 3.24-3.26 (m, 5H), 3.36-3.46 (m, 5H), 3.65-3.69 (m, 1H), 3.86 (d, J = 1 1.9 Hz, 1H), 3.97-4.08 (m, 5H), 6.82-6.84 (m, 2H), 7.10 (d, J = 8.6 Hz, 2H), 7.26 (dd, J = 8.1 and 2.0, 1H), 7.31 (d, J = 2.0 Hz, 1H), 7.33 (d, J= 8.2 Hz, 1H).
ESIMS (m/z): 557.8 (M+l)
EXAMPLE X: Preparation of (2S,3R,4R,5S,6i?)-2-(3-(4-(2-
(sulfamoylamino)ethoxy)benzyl)-4-chlorophenyl)-tetrahydro-6-((3-cyclobutyl ureido) methyl)-2H-pyran-3,4,5-triol

Step 1: Preparation of ((2i?,35,4i?,5JR,65)-6-(4-chloro-3-(4-(2-(sulfamoylamino)ethoxy) benzyl)phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methyl 4- methylbenzenesulfonate
To a solution of (21S,3/?,4i?,55,6/?)-2-(3-(4-(2-(sulfamoylamino)ethoxy)benzyl)-4- chlorophenyl)-tetrahydro-6-(hydroxymethyl)-2H-pyran-3,4,5-triol (630 mg, 1.25 mmol) in 2,6-leutidine (6.2 mL), /?-toluenesulfonylchloride (1200 mg, 6.26 mmol) was added at r.t. The resulting solution was stirred for 16 h while montoring on TLC. On completion, reaction mixture was diluted with ethyl acetate (50 mL), washed with 5% HCl (2 x 20 mL), water (20 mL) and brine (20 mL) successively. The organic layer was dried over anhydrous Na2S0 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :4 MeOH: DCM) to provide the title compound (605 mg, 73 %) as white solid.
ESIMS (m/z): 657.3 (M+l)
Step 2: Preparation of ((2JR,35,4i?,5i?,65)-6-(4-chloro-3-(4-(2-(sulfamoylamino) ethoxy)benzyl)phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methylazide
To a solution of ((2R,3S,4R,5R,6S)-6-(4-chloro-3-(4-(2-
(sulfamoylamino)ethoxy)benzyl) phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2- yl)methyl 4-methylbenzenesulfonate (600 mg, 0.91 mmol) in dry DMF (4.5 mL), sodiumazide (297 mg, 4.56 mmol) and tetrabutylammoniumiodide (34 mg, 0.091 mmol) were added at r.t. The resulting solution was heated to 60 °C while stirring for 16 h. The reaction mixture was diluted with ethyl acetate (50 mL), washed with water (20 mL) and brine (20 mL) successively. The organic layer was dried over anhydrous Na2S0 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :4 MeOH: DCM) to provide the title compound (442 mg, 92 %) as white solid.
ESIMS (m/z): 528.3 (M+l)
Step 3: Preparation of ((2/?,35',4i?,5i?,65)-6-(4-chloro-3-(4-(2-(sulfamoylamino)ethoxy) benzyl)phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methylamine
To a solution of ((2i?,35,4i?,5 ?,6lS)-6-(4-cliloro-3-(4-(2-(sulfamoylamino)ethoxy) benzyl) phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methylazide (440 mg, 0.833 mmol) in THF:water (1 :1, 4 raL), triphenylphosphine (328 mg, 1.24 mmol) was added. The resulting solution was stirred at r.t. for 16 h. The volatiles were evaporated and the residue obtained was purified by column chromatography (silica gel, 1 :4 MeOH: DCM) to provide the title compound (370 mg, 89 %) as white solid.
ESIMS (m/z): 502.7 (M+l)
Step 4: Preparation of (25,3i?,4i?,55,6i?)-2-(3-(4-(2-(sulfamoylamino)ethoxy)benzyl)-4- chlorophenyl)-tetrahydro-6-((3-cyclobutyl ureido) methyl)-2H-pyran-3,4,5-triol
Step 4.1: To a solution of cyclobutylamine (200 mg, 2.81 mmol) in dry DCM (14 mL), carbonyldiimidazole (755 mg, 4.21 mmol) was added at 0 °C. The resulting mixture was stirred at r.t. for 2h. Reaction mixture was diluted with DCM (25 mL) and washed with water (20 mL) and brine (20 mL) successively. Organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo to obtain N-cyclobutyl-lH-imidazoIe- 1 -carboxamide as crude mixture. This was used as such in the next step.
Step 4.2: To a solution of ((2i?,35',4JR,5/?,61S)-6-(4-chloro-3-(4-(2-(sulfamoylamino)ethoxy) benzyl )phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methylamine (120 mg, 0.24 mmol) in dry THF (1.15 mL), N-cyclobutyl-lH-imidazole-1 -carboxamide (39.4 mg, 0.24 mmol) was added at r.t. The resulting mixture was stirred at r.t. for 16 h and the reaction was monitored by TLC. On completion, reaction mixture was diluted with ethyl acetate (20 mL), washed with water (10 mL) and brine (10 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide the title compound (130 mg, 91 %) as white solid.
1H NMR (400 MHz, CD3OD, δ): 1.58-1.62 (m, 4H), 2.19-2.20 (m, 2H), 3.26 (d, J = 8.2 Hz, 1H), 3.29-3.33 (m, 2H), 3.37-3.46 (m, 4H), 3.54 (d, J = 13.4 Hz, 1H), 4.01-4.09 (m, 6H), 6.84-6.86 (m, 2H), 7.10 (d, J = 8.3 Hz, 2H), 7.26-7.27 (m, 2H), 7.35 (d, J = 8.1 Hz, 1H).
ESIMS (m/z): 597.7 (M-l)
EXAMPLE XI: Preparation of (2R,3S,4R,5R,6S)-2-((2,2,2-trifluoroethoxy)methyl)-6-
(3-(4-(4-aminosulfonylaminobutoxy)benzyl)-4-chlorophenyl)-tetrahydro-2H-pyran-
3,4,5-triol

Step 1: Preparation of (2R,3R,4R,5S,6S)-2-(acetoxymethy\)-6-(3-(4-((tert- butyldimethylsilyl)oxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate To a solution of (2 ?,3i?,4i?,5, ,66 -2-(Acetoxymethyl)-6-(4-chloro-3-(4- hydroxybenzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (500 mg, 0.91 mmol) in dry DCM (2.70 mL), triethyl amine (0.32 mL, 2.27 mmol) and 4-dimethylaminopyridine (10 mg, 0.08 mmol) were added at 0 °C followed by the addition of tert- butyldimethylsilylchloride (275 mg, 1.82 mmol). The resulting mixture was stirred at r.t. for 16 h and the reaction was monitored by TLC. On completion, reaction mixture was diluted with DCM (20 mL), washed with water (10 mL) and brine (10 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 acetone: Pet. ether) to provide the title compound (530 mg, 87.74 %) as white solid.
ESI-MS (m/z): 685.8 (M+23)
Step 2: Preparation of (2S,3/?,4i?,55,6i?)-2-(3-(4-((iert-butyldimethylsilyl)oxy)benzyl)-4- chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
Sodium (20 mg) was added to dry methanol (10.93 mL) and stirred till sodium was dissolved. A solution of (2R,3R,4R,5S, 65)-2-(acetoxymethyl)-6-(3-(4-((½rt- butyldimethylsilyl)oxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (2.9 g, 4.37 mmol) in dry methanol (10.9 mL) was added to it and stirred for 1 h at r.t. The volatiles were evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide the title compound (1.83 g, 84.7 %) as white solid.
ESI-MS (m/z): 495.2 (M+l)
Step 3: Preparation of ((27?,31S',4i?,5i?,6iS -6-(3-(4-((fer/-butyldimethylsilyl)oxy)benzyl)-4- chlorophenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methy]-4- methylbenzenesulfonate
To a solution of (25,3i?,4i?,55,6i?)-2-(3-(4-((/eri-butyldimethylsilyl)oxy)benzyl)-4- chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (340 mg, 0.68 mmol) in 2,6-leutidine (4.72 mL), -toluenesulfonylchloride (655 mg, 3.43 mmol) was added at r.t.
The resulting solution was stirred for 16 h while montoring on TLC. On completion, reaction mixture was diluted with ethyl acetate (50 mL), washed with 5% HC1 (2 x 20 mL), water (20 mL) and brine (20 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :9 acetone: DCM) to provide the title compound (400 mg, 89.9 %) as white solid.
ESI-MS (m/z): 667.0 (M+18)
Step 4: Preparation of (25,,3i?,4/?,55',6i?)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- ((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-3,4,5-triol
To a solution of ((2i?,35,4/?,5i?,6 )-6-(3-(4-((½rt-butyldimethylsilyl)oxy)benzyl)-
4-chlorophenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methyl 4- methylbenzenesulfonate (1 g, 1.54 mmol) in 2,2,2-trifluoroethanol (10 mL) was added 60% sodium hydride (616 mg, 15.4 mmol). The reaction was heated to 1 10 °C while stirring for 16 h and monitored by TLC. On completion, reaction mixture was diluted with ethyl acetate (50 mL), washed with water (20 mL) and brine (20 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide the title compound (290 mg, 40.6 %) as white solid.
ESI-MS (m/z): 462.1 (M-l)
Step 5: Preparation of tert-butyl (4-(4-(2-chloro-5-((21S,3 ?,4JR,55,6i?)-3,4,5-trihydroxy-6- ((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)butyl)caribamate To a solution of (25',3^,4 ?,51S',6 ?)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- ((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-3,4,5-triol (150 mg, 0.32 mmol) and 4-((teri-butoxycarbonyl)amino)butyl methanesulfonate (173 mg, 0.65 mmol) in dry DMF (1 mL), cesium carbonate (316 mg, 0.97 mmol) and sodium iodide (73 mg, 0.49 mmol) were added at r.t. and stirred for 48 h. On completion, as monitored by TLC, the resulting mixture was diluted with ethylacetate (20 mL) and washed with water (2 x 10 mL) and brine (10 mL) successively. Organic layer was separated, dried over anhydrous Na2S0 and the volatiles were evaporated in vacuo. The residue was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide title compound (150 mg, 73.2 %) as off white solid.
ESI-MS (m/z): 657.0 (M+23)
Step 6: Preparation of (21S,,35,4i?,5 ?,67?)-2-(3-(4-(4-((teri-butoxycarbonyl)amino) butoxy)benzyl)-4-chlorophenyl)-6-((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran- 3,4,5-triyl triacetate
To a solution of ierf-butyl (4-(4-(2-chloro-5-((25,3.R,4i?,55,6i?)-3,4,5-trihydroxy-6- ((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)butyl)carbamate (360 mg, 0.56 mmol) in dry THF (3 mL), N-ethyldiisopropylamine (0.55 mL, 3.12 mmol) and 4-dimethylaminopyridine (19 mg, 0.16 mmol) were added at 0 °C followed by the addition of aceticanhydride (0.26 mL, 2.76 mmol). The resulting mixture was stirred at r.t. for 1 h and the reaction was monitored by TLC. On completion, reaction mixture was diluted with ethylacetate (20 mL), washed with water (10 mL) and brine (10 mL) successively. The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 3:7 acetone: Pet. ether) to provide the title compound (410 mg, 95.12 %) as white solid.
ESI-MS (m/z): 783.1 (M+23)
Step 7: Preparation of (25,35,4i?,5i?,6/?)-2-(3-(4-(4-aminobutoxy)benzyl)-4- chlorophenyl)-6-((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2,2,2-trifluoroacetate
To a solution of (25,3^,4 ?,5^,6i?)-2-(3-(4-(4-((½rt-butoxycarbonyl)amino) butoxy)benzyl)-4-chlorophenyl)-6-((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran- 3,4,5-triyl triacetate (400 mg, 0.53 mmol) in dry DCM (1.05 mL), trifluoroaceticacid (1.57 mL) was added at 0 °C. The resulting mixture was stirred while gradually raising the temperature to r.t. for 1 h. Volatiles were evaporated in vacuo and the residue obtained was washed with diethyl ether (2 x 20 mL) to provide title compound (410 g, 100 %) as off-white solid.
ESI-MS (m/z): 660.5 (M+l , for free amine)
Step 8: Preparation of (25,35,4ii,5JR,6i?)-2-(4-chloro-3-(4-(4-(sulfamoylamino)butoxy) benzyl)phenyl)-6-((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate To a solution of (2S,35,4R,5i?,6R)-2-(3-(4-(4-aminobutoxy)benzyl)-4- chlorophenyl)-6-((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 2,2,2-trifluoroacetate (250 mg, 0.323 mmol) in dry dioxane (1.6 mL), triethylamine (0.14 mL, 0.97 mmol) was added at 0 °C and stirred while gradually raising the temperature to r.t. Sulfamide (47 mg, 0.48 mmol) was added and the resulting mixture was refluxed for 5 h. Dioxane was evaporated in vacuo and the residue obtained was diluted with ethylacetate
(50 mL). Organic layer was waned with water (20 mL) and brine (20 mL) successively. Ethylacetate layer was dried over anhydrous Na2S04 ,and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 acetone: Pet. ether) to provide the title compound (90 mg, 37.8 %) as white solid.
ESI- S (m/z): 739.9 (M+l)
Step 9: Preparation of (2/?,3lS',4v,?,57?,65)-2-((2,2,2-trifluoroethoxy)methyl)-6-(3-(4-(4- aminosulfonylaminobutoxy)benzyl)-4-chlorophenyl)-tetrahydro-2H-pyran-3,4,5-triol
To a solution of (21S,35,4JR,5 ?,67?)-2-(4-chloro-3-(4-(4-(sulfamoylamino) butoxy)benzyl)phenyl)-6-((2,2,2-trifluoroethoxy)methyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (80 mg, 0.11 mmol) in THF (0.36 mL), methanol (0.54 mL) and water (0.18 mL) was added lithium hydroxide monohydrate (9 mg, 0.22 mmol) at 0 °C. The reaction was stirred at r.t. for 1 h. Volatiles were evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to provide the title compound (40 mg, 60.6 %) as white solid.
Ή NMR (400 MHz, CD3OD, 8): 1.70-1.84 (m, 4H), 3.08 (t, J = 7.0 Hz, 2H), 3.25-3.27 (m, 1H), 3.40-3.47 (m, 3H), 3.82 (dd, J = 1 1.6 and 4.9 Hz, 1H), 3.90-4.01 (m, 7H), 4.06 (d, J= 9.5 Hz, 1H), 6.79-6.81 (m, 2H), 7.08 (d, J= 8.6 Hz, 2H), 7.21 (dd, J = 8.2 and 2.1 Hz, 1H), 7.24 (d, J = 1.9 Hz, 1H), 7.34 (d, J= 8.2 Hz, 1H).
ESI-MS (m/z): 613.9 (M+l)
EXAMPLE XII: Preparation of N-(N-(2-(4-(2-chloro-5-((25,3R,4R,5S,6 ?)-3,4,5- trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2- yl)benzyI)phenoxy)ethyI)sulfamoyl) acetamide

Step 1: Preparation of (2i?,37?,4i?,55,65)-2-(acetoxymethyl)-6-(3-(4-(2-((N- acetylsulfamoyl) amino)ethoxy) benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (2i?,3ii,4i?,51S,61S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2 (sulfamoylamino) ethoxy)benzyl) phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (170 mg, 0.253 mmol) in 1 ,4-dioxane ( 0 mL) acetyl chloride (0.06 mL, 0.89 mmol) was added
at r.t. The reaction mixture was refluxed at 100 °C temperature for 24 h under argon. After completion of reaction, as confirmed by TLC, the reaction mixture was diluted with EtOAc (150 mL) and washed with water (5 x 10 mL). The organic layer was dried over anhydrous Na2S04 and volatiles were removed in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :24 MeOH: DCM) to afford title compound (100 mg, 55.0%) as a white solid.
ESI-MS (m/z): 731.0 (M+18)
Step 2: Preparation of N-(N-(2-(4-(2-chloro-5-((25,3 ?,4i?,55,6i?)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)sulfamoyl)acetamide To a solution of (2/?,3R,4R,55,65)-2-(acetoxymethyl)-6-(3-(4-(2-((^- acetylsulfamoyl) amino)ethoxy) benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (150 mg, 0.21 mmol) in MeOH:THF:Water (3:2: 1, 2.1 mL) was added lithium hydroxide monohydrate (8.8 mg, 0.210 mmol) at 0 °C . The reaction mixture, was stirred at r.t. for 1 h. After completion of reaction, as confirmed by TLC, the reaction mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to afford the title compound (95mg, 83%) as white solid. Ή NM (400 MHz, CD3OD, 8): 1.93 (s, 3H), 3.25-3.29 (m, 2H), 3.36-3.38 (m, 2H), 3.40-3.44 (m, 1H), 3.5 (t, J = 5.5 Hz, 2H), 3.65 - 3.69 (m, 1H), 3.84-3.87 (m, 1H), 3.98- 4.00 (t, J = 5.5Hz, 2H), 4.03 (d, J = 8.5 Hz, 1H), 4.08 (d, J = 9.5 Hz, 1H), 6.81-6.83 (m, 2H), 7.08-7.10 (m, 2H), 7.26 (dd, J= 8.2 and 2.1 Hz, 1H), 7.30-7.34 (m, 2H).
ESI-MS (m/z): 568.2 (M+23)
EXAMPLE XIII: Preparation of N-(2-(4-(2-chloro-5-((25,3R,4R,5S,6R)-3,4,5- trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)-
N^V^V-trimethylsulphamide

Step 1: Preparation of (2J ,3i?,4JR,5S,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2-((N,N- dimethylsulfamoyl)(methyl)amino)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (2i?,3i?,4i?,5S,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2 (sulfamoylamino)ethoxy)benzyl) phenyl )tetrahydro-2H-pyran-3,4,5-triyl triacetate (1 10 mg, 0.16 mmol) in dry DMF (3 mL), cesium carbonate (213 mg, 0.67 mmol) was added followed by the addition of methyl iodide (0.03 mL, 0.49 mmol) at 0 °C . The reaction was stirred at r.t. for 24 h. After completion, as confirmed by TLC, the reaction mixture was diluted with ethylacetate (150 mL) and washed with water (2 x 20 mL). The organic layer was dried over anhydrous Na2S04 volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 4:6 acetone: Pet. ether) to afford title compound as a white solid (105mg, 98.32%).
ESI-MS (m/z): 713.6 (M+l)
Step 2: Preparation of N-(2-(4-(2-chloro-5-((2S,3i?,4i?,55,6/?)-3,4,5-trihydroxy-6-
(hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)-N,7V,7V- trimethylsulphamide
The title compound (52mg, 64.9 %) was synthesized following the procedure reported for the synthesis of N-(N-(2-(4-(2-chloro-5-((25,3i?,4i?,55,6i?)-3,4,5-trihydroxy-6- (hydroxymethyl) tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)sulfamoyl)acetamide starting with (2i?,3i?,4JR,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2-((7V,N- dimethylsulfamoyl)(methyl) amino)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (105mg, 0. 47mmol).
Ή NMR (400 MHz, CD3OD, 6): 2.77 (s, 6H), 2.92 (s, 3H), 3.25 (s, 1H), 3.3 (d, J = 7.1 Hz, 2H), 3.41-3.44 ( m, 3H), 3.53 (t, J = 5.35 Hz, 2H), 3.56-3.69 (m, 1H), 3.86 (d, J = 1 1.6 Hz, 1H ), 4.02 (d, J = 8.2 Hz, 2H), 4.06-4.1 1 (m, 3H), 6.83 (d, J = 8.7 Hz, 2H), 7.10 (d, J= 8.6 Hz, 2H ), 7.25-7.27 (dd, J= 8.2 and 2.0 Hz, 1H), 7.31-7.34 (m, 2H).
ESI-MS (m/z): 567 (M+23), 545.9 (M+l)
EXAMPLE XIV: Preparation of N-(2-(4-(2-chloro-5-((25,3R,4R,5S,6R)-3,4,5- trihydroxy-6-(piperidin-l-ylmethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl) methanesulfonamide

To a solution of (2 ,3#,4^5S,65)-2-(acetoxymethyl)-6-(3-(4-(2- aminoethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyltriacetate 2,2,2- trifiuoroacetate (700 mg, 0.99 mmol) in dry DCM (5 mL), triethylamine (0.41 mL, 2.97 mmol) followed by methanesulfonylchloride (0.12 mL, 1.48 mmol) were added at 0 °C. The resulting mixture was stirred at the same temperature for lh. It was diluted with DCM (20 mL), washed with water (20 mL) and brine (20 mL) successively. Organic layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 acetone: Pet. ether) to provide the title compound (500 mg, 75.32 %) as white solid.
ESIMS (m/z): 669.2 (M-l)
Step 2: Preparation of N-(2-(4-(2-chloro-5-((25,3^,4^,5^,6^)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)methanesulfonamide The title compound (350 mg, 9.45 %) was synthesized following the procedure reported for the synthesis of (25,,3^,4 ?,5.S,,6^)-2-(3-(4-(2- (sulfamoylamino)ethoxy)benzyl)-4-chlorophenyl)-tetrahydro-6-(hydroxymethyl)-2H- pyran-3,4,5-triol starting with (2i?,3i?,4 ?,55,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- (methylsulfonamido)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (500 mg, 0.746 mmol).
ESIMS (m/z): 524.9 (M+23)
Step 3: Preparation of ((2i!?,31S',4^,5^,6l )-6-(4-chloro-3-(4-(2-(methylsulfonamido) ethoxy)benzyl)phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-yl)methyl-4- methylbenzenesulfonate
The title compound (1.2 g, 92.3 %) was synthesized following the procedure reported for the synthesis of ((2i?,35,4i?,5i?,65)-6-(4-chloro-3-(4-(2- (sulfamoylamino)ethoxy)benzyl)phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2- yl)methyl 4-methylbenzenesulfonate starting with N-(2-(4-(2-chloro-5-((2S,3R,4X,5S,6R)- 3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2- yl)benzyl)phenoxy)ethyl)methanesulfonamide (1.0 g, 0.002 mol).
ESIMS (m/z): 678.2 (M+23), 656.2 (M+l).
Step 4: Preparation of N-(2-(4-(2-chloro-5-((25,3 ?,4i?,55,6i?)-3,4,5- trihydroxy-6- (piperidin- 1 -ylmethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)
methanesulfonamide
To a solution of ((2R,3S,4R,5/?,65)-6-(4-chloro-3-(4-(2-
(methylsulfonamido)ethoxy) benzyl)phenyl)-3,4,5-trihydroxytetrahydro-2H-pyran-2- yl)methyl 4-methylbenzenesulfonate (140 mg, 0.24 mmol) in dry DMF (5 mL) was added triethylamine (0.2 mL, 1.42 mmol) and piperidine (60 mg, 0.71 mmol) at r.t. The reaction mixture was stirred at 80 °C for 36 h under argon. After completion of reaction, as confirmed by TLC, the reaction mixture was diluted with ethylacetate (150 mL) and washed with water (5 x 20 mL). The organic layer was dried over anhydrous Na2S04 and the volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :9 MeOH: DCM) to afford title compound (60 mg, 44.5%) as a white solid.
1H NMR (400 MHz, CD3OD, 5): 1.45-1.48 (m, 2H), 1.58-1.62 (m, 4H), 2.70-2.75 (m, 5H), 2.97 (s, 3H), 3.02 (dd, J = 13.6 and 3.0 Hz, 1H), 3.21-3.26 (m, 2H), 3.41-3.46 (m,3H),3.59-3.65 (m,lH), 4.02-4.04 (m, 4H), 4.10 (d, J = 9.5 Hz, 1 H), 6.83-6.86 (m, 2H), 7.09-7.1 l ( m, 2H ), 7.1 1-7.25 (m, 2H), 7.33 (d, J= 8.2 Hz, 1H)
ESI-MS (m/z): 570.8 (M+l)
EXAMPLE XV: Preparation of l-(2-(4-(2-chloro-5-((25,3 ?,4/?,55,6/?)-3,4,5-trihydroxy- 6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)-4-phenyl- 1 ,4- azaphosphinane 4-oxide
 Step 1: Preparation of (2-bromoethoxy)(iert-butyl)dimethylsilane
Step 1: Preparation of (2-bromoethoxy)(iert-butyl)dimethylsilane
To a solution of 2-bromoethanol (3.4 mL, 48.34 mmol) in dry DCM (25 mL) imidazole (9.86 g, 143 mmol) and /eri-butylchlorodimethylsilane (10.9gm, 72.52 mmol) were added at 0 °C. The reaction mixture was stirred at r.t. for 24 h. After completion of reaction, as confirmed by TLC, the reaction mixture was diluted with ethylacetate (350 mL) and washed with water (4 x 20 mL). The organic layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 1 :49 EtOAc: Pet. ether) to afford title compound (204 mg, 77.5 %) as liquid.
ESI-MS (m/z): 239.1 (M+l).
Step 2: Preparation of (2i?,3i?,4i?,55,65)-2-(acetoxymethyl)-6-(3-(4-(2-((tert- butyldimethylsilyl)oxy)ethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (2 ?,3R,4 ?,5S,65)-2-(Acetoxymethyl)-6-(4-chIon)-3-(4- hydroxybenzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (500 mg, 0.91 mmol) in dry DMF (10 mL), was added (2-bromoethoxy)(½rt-butyl)dimethylsilane (436 mg, 1.82 mmol) and cesium carbonate (890 mg, 2.73 mmol). The reaction was stirred at r.t. for 36 h. After completion of reaction, as confirmed by TLC, the reaction mixture was diluted with ethylacetate (150 mL) and washed with water (3 x 10 mL). The organic layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:3 Acetone: Pet. ether) to afford title compound (392 mg, 60.85%) as white solid.
ESI-MS (m/z): 708(M+l )
Step 3: Preparation of (2 ?,3^,4i?,5^,65)-2-(acetoxymethyI)-6-(4-chloro-3-(4-(2- hydroxyethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution (2i?,3i?,4 ?,55,65 -2-(acetoxymethyl)-6-(3-(4-(2-((tert- butyldimethylsilyl)oxy)ethoxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (2.59 g, 3.66 mmol) in THF:Water (1 : 1 , 36 mL) was added acetic acid (54 mL) at 0 °C. The reaction mixture was stirred at r.t. for 2 h. After completion of reaction, as confirmed by TLC, the reaction mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 2:3 EtOAc: Pet ether) to afford title compound (1.8g, 82 %) as a liquid.
ESI-MS (m/z): 593.7(M+1)
Step 4: Preparation of (2/?,3i?,4 ?,55',65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- (tosyloxy)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of (2R,3R,4R,5S,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- hydroxyethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (290 mg,0.49 mmol) in 2,6-leutidine (3.4 mL) was added p- toluenesulfonyichloride (558 mg, 2.94 m.mol) at 0 °C. The reaction mixture was stirred at r.t. for 24 h. After completion of reaction, as confirmed by TLC, the reaction mixture was diluted with ethyl acetate (150 mL) and washed with 2N HC1 (3 x 20 mL). The organic layer was dried over anhydrous Na2S0 and volatiles were evaporated in vacuo. The residue obtained was purified by
column chromatography (silica gel, 1 : 1 EtOAc: Pet. ether) to afford title compound (130 mg, 35%) as white solid.
ESI-MS (m/z): 769.9(M+23)
Step 5: Preparation of (2i?,3i?,4^,55,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2-(4- oxido-4-phenyl-l ,4-azaphosphinan-l-yl)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5- triyl triacetate
To a solution of (2i?,3/?,4/?,55,65)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2- (tosyloxy)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (130 mg, 0.17 mmol) in dry DMF (8 mL), triethylamine (0.072 mL, 0.52 mmol) and 4-phenyl-l,4- azaphosphinane 4-oxide (68mg, 0.346 m.mole, prepared according to the procedure given in WO2008090570 and WO2009116090) were added at r.t. The reaction mixture was stirred at 80 °C for 36 h. After completion of reaction, as confirmed by TLC, the reaction mixture was diluted with EtOAc (150 mL) and washed with water (3 x 20 mL). The organic layer was dried over anhydrous Na2S04 and volatiles were evaporated in vacuo. The residue obtained was purified by column chromatography (silica gel, 2:48 MeOH: DCM) to afford title compound (65mg, 63.1%) as a white solid.
ESI-MS (m/z): 771.7 (M+l)
Step 6: Preparation of l-(2-(4-(2-chloro-5-((25,3^,4 ?,55,6i?)-3,4,5-trihydroxy-6- (hydroxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)phenoxy)ethyl)-4-phenyl-l ,4- azaphosphinane 4-oxide
To a solution of 2 ?,3^,4JR,51S',6k5,)-2-(acetoxymethyl)-6-(4-chloro-3-(4-(2-(4-oxido- 4-phenyl- 1 ,4-azaphosphinan- 1 -yl)ethoxy)benzyl)phenyl)tetrahydro-2H-pyran-3 ,4,5-triyl triacetate (93 mg, 0.13 mmol) in MeOH:THF:Water (3:2:1 , 1.26 mL) was added lithium hydroxide monohydrate (5.07 mg, 0.13 mmol) at 0 °C . The reaction mixture was stirred at r.t. for 1 h. After completion of reaction, as confirmed by TLC, the reaction mixture was evaporated in vacuo and the residue obtained was purified by column chromatography (silica gel, 3:7 MeOH: DCM) to afford title compound (58 mg, 80.3%) as off white solid. Ή NMR (400 MHz, CD3OD, 6): 2.04-2.05 (m, 2H), 2.37-2.38 (m, 2H), 2.96 (t, J = 5.3 Hz, 2H), 3.03-3.09 (m, 2H), 3.1 1-3.13 (m, 1H), 3.16-3.19 (m, 1H), 3.22-3.25 (m, 1H), 3.34-3.37 (m, 2H), 3.38-3.42 (m, 1H), 3.65-3.69 (m,lH), 3.84-3.87 (m, 1H), 3.97-4.03 (m, 2H), 4.06-4.08 (m, 1H), 4.09-4.12 (m, 1H), 6.82-6.86 (m, 2H), 7.1 (d, J = 8.7 Hz, 2H), 7.27 (dd, J= 8.2 and 2.1 Hz, 1Η),7.31-7.34 (m, 2H),7.54-7.64 (m, 3H),7.78-7.83 (m, 2H). ESI-MS (m/z): 602.5 (M+l)
The compounds listed in Tables 2 to 5 were prepared essentially following the procedures described for Examples I to XIV:
Table 2


Table 3


Table 4


Table 5

 The SGLT inhibitory effects of the compounds of the present invention were demonstrated by following test procedures. In vitro Studies
The SGLT inhibitory effects of the compounds of the present invention were demonstrated by following test procedures. In vitro Studies
Preparation of mouse SGLT-2 expressing cells
Full-length mouse SGLT-2 cDNA was amplified from C57BL/6J mouse kidneys and introduced in the pcDNA3.1(+) expression vector (Invitrogen, Inc.) and propagated in Escherichia coli strain DH5a using Luria-Bertani (LB) medium containing ampicillin. Mouse SGLT-2 recombinant expression plasmid DNA was transfected into CHO-K1/HEK cells (American Type Culture Collection) using Superfect Transfection Reagent according to a manufacturer suggested protocol. Stably transfected cells were selected using G418 antibiotic selection pressure.
Methyl-a-D-[U-, C] Glucopyranoside uptake assay for SGLT-2
Cells expressing mSGLT-2 were seeded on 96-well tissue culture plates (Greiner, Inc.) in RPMI containing 10% FBS and 400μg/mL G418 (0.8 x 105 cells per well in 200μί medium) and incubated at 37 °C under 5% carbon dioxide for 24 h prior to the assay. Cells were washed twice with 200μί of either sodium buffer (140 mM NaCl, 4.7 mM KC1, 2.2 mM CaCl2, 1.2 mM MgCl2, 10 mM Tris/Hepes, pH 7.4) or sodium-free buffer (137 mM N-methyl-glucamine, 4.7 mM KC1, 2.2 mM CaCl2, 1.2 mM MgCI2, 10 mM Tris/Hepes, pH 7.4). Reaction mixture containing test compounds diluted in assay buffer, O. lmM unlabeled Methyl-a-D-glucopyranoside and ^Ci/well methyl-a-D-[U- 14C] glucopyranoside (American Radiochemicals) was added per well of a 96-well plate and incubated at 37 °C for either 1 h. Cells were washed thrice with 200μί of wash buffer (140 mM NaCl, 4.7 mM KC1, 2.2 mM CaCl2, 1.2 mM MgCl2, 10 mM Tris/Hepes, pH 7.4 containing 500μΜ phlorizin) and lysed using 50 ί of 0.25N NaOH. Methyl-oc-D-[U- 14C]glucopyranoside uptake was quantitated using a Top count scintillation counter (PerkinElmer, Inc.). All the test compounds were assayed in triplicates.
Table 6
S. No. 1 Compound No. % inhibition of Methyl-a-D-[U- C] Glucopyranoside uptake
(CHO-K1/HE1 : cells) mediated by SGLT-2
0.1 nM InM ΙΟηΜ ΙΟΟηΜ ΙΟΟΟηΜ
1 13 N.S. N.S. 71.66 72.25 65.50
(HEK cells) (HEK cells) (HEK cells)
2 18 N.E. 19.41 63 N.S. N.S.
(CHO-Kl (CHO-Kl (CHO-Kl
cells) cells) cells)
3 20 N.S. 77.90 79.63 83.78 N.S.
(HEK cells) (HEK cells) (HEK cells)
4 23 N.S. 47.29 71.32 83.15 N.S.
(HEK cells) (HEK cells) (HEK cells)
5 24 N.E. 19.54 56.04 N.S. N.S.
(CHO-Kl (CHO-Kl (CHO-Kl
cells) cells) cells)
6 25 N.S. N.S. 68.60 74.12 70.92
(HEK cells) (HEK cells) (HEK cells)
7 29 N.S. N.S. 38.72 67.52 66.50
(HEK cells) (HEK cells) (HEK cells)
8 31 N.S. N.S. 56.35 61.77 62.98
(HEK cells) (HEK cells) (HEK cells)
N.E. = Not Effective; N.S. = Not Screened
Preparation of human SGLT-1 expressing cells
Full-length human SGLT-1 cDNA in the pCMV-XL-Neo expression vector was obtained from Origene Corporation and propagated in Escherichia coli strain DH5a using Luria- Bertani (LB) medium containing ampicillin. Human SGLT-1 expression plasmid DNA was transfected into CHO-Kl cells (American Type Culture Collection) using Superfect Transfection Reagent according to a manufacturer suggested protocol. Stably transfected cells were selected using G418 antibiotic selection pressure.
Methyl-a-D-[U- C] Ghicopyranoside uptake assay for SGLT-1
Cells expressing hSGLT-1 were seeded on 96-well tissue culture plates (Greiner, Inc.) in
RPMI containing 10% FBS and 800μg/ml G418 (0.8 x 105 cells per well in 200μί medium) and incubated at 37 °C under 5% carbon dioxide for 24 h prior to the assay. Cells were washed twice with 200μί of either sodium buffer (140 mM NaCl, 4.7 mM KCl, 2.2 mM CaCl2, 1.2 mM MgCl2, 10 mM Tris/Hepes, pH 7.4) or sodium-free buffer (137 mM
N-methyl-glucamine, 4.7 mM KCl, 2.2 mM CaCl2, 1.2 mM MgCl2, 10 mM Tris/Hepes, pH 7.4). Reaction mixture containing test compounds diluted in assay buffer, ImM unlabeled Methyl-a-D-glucopyranoside and ^Ci/well methyl-a-D-[U-
C]glucopyranoside (American Radiochemicals) was added per well of a 96-well plate and incubated at 37 °C for either 1 h. Cells were washed thrice with 200μί of wash buffer (140 mM NaCl, 4.7 mM KC1, 2.2 mM CaCl2, 1.2 raM MgCl2, 10 mM Tris/Hepes, pH 7.4 containing 500μΜ phlorizin) and lysed using 50μί of 0.25N NaOH. Methyl-ct-D-[U- l4C]glucopyranoside uptake was quantitated using a Top count scintillation counter (PerkinElmer, Inc.). All the test compounds were assayed in triplicates.
Table 7

In vivo Studies
Estimation of urinary glucose in C57/BL 6J mice
C57/BL 6 J mice were fasted 4 h before drug treatment. Fasted mice were weighed and randomized into different groups based on their body weight (n=6). At time To test compounds and standard compounds suspended in 0.25% CMC were administered to respective groups and kept in metabolic cages (6 mice/ cage) after dosing. Animals were fed after drug administration and urine was collected over a period of 24 h. Urinary volume was measured and urinary glucose was estimated using Merckotest Glucose Reagent (Merck, Inc.).
Table 8
W

Claims
1. A compound of Formula I,

Formula I
or its pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, wherein:
ring A represents aryl;
ring B represents either aryl or heteroaryl;
U, V and W are independently selected from -OH, hydrogen, halogen, Ci_i2alkoxy, -CN, -(CH2)nNR8 9, -OR8, -C(=Y)OR8 or -C(=Y)NR8R9; provided that atleast two groups out of U, V and W represent -OR8;
Y represents either O or S;
R7 is selected from halogen, Ci-i2alkyl, C2-i2alkenyl, C2-i2alkynyl, C|.i2alkylcarbonyl, CM2 alkoxycarbonyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)nRe, -CN, -N02, - NR8R9, -N3, -CR8(=NOR9), -OH, -OR8, -CH2OH, -C(=Y)R8, -C(=Y)OR8, -C(=Y)SR8, - C(=Y)NR8R9, -OC(=Y)R8, -OC(=Y)OR8, -OC(=Y)NR8R9, -OP(=0)R8R9, -(CH2)n- heterocyclyl, -(CH2)n-NR8R9, -(CH2)n-N3, -(CH2)n-NCS, -(CH2)n-S(0)dR8, -(CH2)n- S(0)dNR8R9, -(CH2)n-P(=0)R8R9, -(CH2)n -OP(=0)R8R9, -(CH2)n-NR8C(=Y)R9, -(CH2)n- NR8C(=Y)OR9, -(CH2)n-NR10C(=Y)NR8R9, -(C¾)n-NR8S(0)dR9 or -(CH2)n- NHP(=0)R8R9 ; each of which may optionally be substituted at any available position by one or more substituents selected from Rn;
R1 , R2, R3 and R4 are independently selected from hydrogen, halogen, Ci-i2alkyl, C -i2 alkenyl, C2_i2alkynyl, Ci.^haloalkyl, C2.i2haloalkenyl, C2_i2haloalkynyl, Ci-nalkoxy, C\_ i2 haloalkoxy, C]_6alkoxyCi_ alkyl,  Ci_i2alkylcarbonyl, Cpi2 alkoxycarbonyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, cycloalkenyl, cycloalkynyl, -(CH2)„-heterocyclyl, -(CH2)n-aryl, -(CH2)„-heteroaryl, - CN, -N02, -NR12R13, -(CH2)nNR12R13, -N3, -NCO, -(CH2)„N3, -(CH2)„ NCS, - CRl 2(=NOR13), -NR1 NR12R13, oxo, -OR12, -SR-12, -(CH2)nYR12, -S(0)dR12, - S(0)dNR, Ri3, -(CH2)nS(0)dRi2, -(CH^SCO^NR^R13, . -P(=0)R, R13, - (CH2)„P(=0)R,2R13, ' -C(=Y)R12, -C(=Y)OR12, -C(=Y)SR12, -C(=Y)NRl2R13, - (CH2)nC(=Y)R12, -(CH2)nC(-Y)OR12, -(CH2)nC(=Y)NR12R13, -(CH2)n-C(=Y)SR12, - OC(=Y)R12, -OC(=Y)OR12, -OC(=Y)NR12R13,-OP(=0)R12R13, -(CH2)nOC(=Y)R12 , - (CH2)nOC(=Y)OR12, -(CH2)nOC(=Y)NR,2R13, -(CH2)nOP(=0)R12R13, -N(R12)C(=Y)R13, -N(R12)C(=Y)OR13, -N(R14)C(=Y)NR12R13, -NRl2S(0)dR13, -NHP(=0)R12R13, - (CH2)nNR12C(=Y)R13, -(CH2)nNR, C(=Y)OR13, -(CH2)nNR14C(=Y)NR12R13, - (CH2)nNR12S(0)dR13 or -(CH2)nNHP(=0)R1 R13; each of which may optionally be substituted at any available position by one or more substituents selected from R11;
Ci_i2alkylcarbonyl, Cpi2 alkoxycarbonyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, cycloalkenyl, cycloalkynyl, -(CH2)„-heterocyclyl, -(CH2)n-aryl, -(CH2)„-heteroaryl, - CN, -N02, -NR12R13, -(CH2)nNR12R13, -N3, -NCO, -(CH2)„N3, -(CH2)„ NCS, - CRl 2(=NOR13), -NR1 NR12R13, oxo, -OR12, -SR-12, -(CH2)nYR12, -S(0)dR12, - S(0)dNR, Ri3, -(CH2)nS(0)dRi2, -(CH^SCO^NR^R13, . -P(=0)R, R13, - (CH2)„P(=0)R,2R13, ' -C(=Y)R12, -C(=Y)OR12, -C(=Y)SR12, -C(=Y)NRl2R13, - (CH2)nC(=Y)R12, -(CH2)nC(-Y)OR12, -(CH2)nC(=Y)NR12R13, -(CH2)n-C(=Y)SR12, - OC(=Y)R12, -OC(=Y)OR12, -OC(=Y)NR12R13,-OP(=0)R12R13, -(CH2)nOC(=Y)R12 , - (CH2)nOC(=Y)OR12, -(CH2)nOC(=Y)NR,2R13, -(CH2)nOP(=0)R12R13, -N(R12)C(=Y)R13, -N(R12)C(=Y)OR13, -N(R14)C(=Y)NR12R13, -NRl2S(0)dR13, -NHP(=0)R12R13, - (CH2)nNR12C(=Y)R13, -(CH2)nNR, C(=Y)OR13, -(CH2)nNR14C(=Y)NR12R13, - (CH2)nNR12S(0)dR13 or -(CH2)nNHP(=0)R1 R13; each of which may optionally be substituted at any available position by one or more substituents selected from R11;
L is selected from O, S, SO, S02, -C(=0)-, -(CH2)n-, -C(=CH2)-, 1 ,1-cyclopropylene, - - NR - or -(C(R )2)m- ; each methylene group may optionally be substituted with one or more substituents independently selected from halogen, hydroxy, oxo, -C(=0)0-, - C(=0)NR16-, Ci-i2alkyl, Ci-f 2alkoxy, -C3-2ocycloalkyl or -C3-2ocycloalkoxy ;
E can be absent or is selected from CH2, O, S or NR16;
G can be absent or is selected from Q.^alkylene, C2.i2alkenylene, C2-i2alkynylene, Cn2 alkylenecarbonyl, C3-20cycloalkylene, heterocyclyl, aryl, heteroaryl, -NR15-, - (CH2)nNR15-, -(CH2)nS(0)d-, -(CH2)nS(0)d NR15-, -(CH2)nP(=0)R15-, -C(=Y)-, C(=Y)NR15-, -(CH2)nC(=Y)-, -(CH2)nC(=Y)NR15-, -(CH2)nOC(=Y)- ,
(CH2)nOP(=0)R15- or -(CH2)nNR15S(0)d-; each of which may optionally be substituted at any available position by one or more substituents selected from R11;
R5 and R6 are independently selected from hydrogen, CM2alkyl, -S(0)dRa, -S(0)dNRaRb or -P(=0)RaRb; each of which may optionally be substituted at any available position by R1 1; wherein R and Rb can be joined together to form a monocyclic or polycyclic ring, which may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R1 1 ;
or
R5 and R6 are joined together along with the nitrogen atom to which they are attached to form a monocyclic or polycyclic ring, which contains atleast one phosphorus atom and
* may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R1 1 ;
provided that
(a) both R5 and R6 can not be hydrogen at the same time
(b) both R5 and R6 can not be alkyl at the same time
(c) R5 and R6 can not be a combination of hydrogen and alkyl at the same time (d) when E and G are absent and R5 is hydrogen then R6 can not represent -S(0)dRa
(e) when R7 represents Ci-i2alkyl, C2-i2alkenyl, C2-]2alkynyl, -CH2OH or -(CH2)„Re, wherein n is not equal to zero; one of R5 and R6 represents -H or Ci-6alkyl and the other represents -S(0)dRa, wherein d represents 1 or 2; then Ra can not be C . 6alkyl, C2- alkenyl, C2-6alkynyl, aryl or heteroaryl;
R8, R9, R10, R12, R13, R14 and R15 are independently selected from hydrogen, halogen, Q. i2alkyl, C2.i2alkenyl, C2-i2alkynyl, Ci-i2alkylcarbonyl, Ci.i2alkoxycarbonyl, C3- 2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, -(CH2)n-heterocyclyl, - (CH2)n-aryl, -(CH2)n-heteroaryl, -CN, -N02, -NRaRb, -(CH2)nNRaRb, -N3, -NCS, - (CH2)„N3, -(CH2)n NCS, -CRa(=NORb), -NRcNRaRb, -0Ra, -SRa, -(CH2)nYRa, -S(0)dRa, -S(0)d NRaRb, -(CH2)nS(0)dRa, -(CH2)n S(0)d NRaRb, -P(=0)RaRb, -(CH2)n P(=0)RaRb, -C(=Y)Ra, -C(=Y)ORa, -C(=Y)SRa, -C(=Y)NRaRb, -(CH2)„C(=Y)Ra, - (CH2)nC(=Y)OR\ -(CH2)nC(=Y)NRaRb, -(CH2)„-C(=Y)SRa, -OC(=Y)Ra, -OC(=Y)ORa, - OC(=Y)NRaRb, -OP(=0)RaRb, -(CH2)nOC(=Y)Ra , -(CH2)nOC(=Y)ORa, - (CH2)nOC(=Y)NRaRb, -(CH2)n0P(O)RaRb, -N(Ra)C(=Y)Rb, -N(Ra)C(=Y)ORb, - N(R°)C(=Y)NRaRb, -NRaS(0)d Rb, -NHP(=0)RaRb, -(CH2)nNRaC(=Y)Rb, - (CH2)nNRaC(=Y)ORb, -(CH2)nNRcC(=Y)NRaRb, -(CH2)nNRaS(0)dRb or -
(CH2)nNHP(=0)RaRb; each of which may optionally be substituted at any available position by one or more substituents selected from Ci-^alkyl, C2-i2alkenyl, C2-i2alkynyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, halogen , -CN, -N02 orNH2; or
R8 and R9 are joined together to form a monocyclic or polycyclic ring, which may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R1 1 ;
R12 and R13 are joined together to form a monocyclic or polycyclic ring, which may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R1 ';
R1 1 is selected from hydrogen, halogen, Ci-i2alkyl, C2-i2alkenyl, C2_i2alkynyl, C)_ )2haloalkyl, C2-i2 haloalkenyl, C2-i2haloalkynyl, Ci_i2alkoxy, C1 -]2haloalkoxy, C\. 6alkoxyCi-6alkyl, Ci-6alkoxyCi_6alkoxyC1-3alkyl, Ci_i2alkylcarbonyl, C\. i2alkoxycarbonyl, C3.2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, - (CH2)„-heterocyclyl, -(CH2)n-aryl, -(CH2)„-heteroaryl, -CN, -N02, -NRaRb, - (CH2)„NRaRb,-N3, -NCS, -(CH2)„N3, -(CH2)„ NCS, -CRa (=NORb), -NR°NRaRb, -ORa, - SRa, -(CH2)nYRa, -S(0)dRa, -S(0)d NRaRb, -(CH2)nS(0)dRa, -(CH2)nS(0)dNRaRb, - P(=0)RaRb, -(CH2)nP(=0) RaRb, -C(=Y)Ra, -C(=Y)ORa, -C(=Y)SRa, -C(=Y)NRaRb, - (CH2)nC(=Y)Ra, -(CH2)nC(=Y)ORa, -(CH2)nC(=Y)NRaRb, -(CH2)n-C(=Y)SRa, - OC(=Y)R\ -OC(=Y)ORa, -OC(=Y)NRaRb, -OP(=0)RaRb, -(CH2)nOC(=Y)Ra , - (CH2)nOC(=Y)OR\ -(CH2)nOC(=Y)NRaRb, -(CH2)nOP(=0)RaRb, -N(Ra)C(=Y)Rb, - N(Ra)C(=Y)ORb, -N(Rc)C(=Y)NRaRb, -NRaS(0)d Rb, -NHP(=0) RaRb, - (CH2)nNRaC(=Y)Rb, -(CH2)nNRaC(=Y)ORb, -(CH2)nNR°C(=Y)NRaRb, (CH2)nNRaS(0)dRb or -(CH2)nNHP(=0) RaRb; each of which may optionally be substituted at any available position by one or more substituents selected from Ci_ i2alkyl, C2-12alkenyl, C2-i2alkynyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, , -CN, - N02 orNH2;
R16 is selected from hydrogen, Ci-i2alkyl, C2-i2alkenyl, C2-|2alkynyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -CRa(=NORb), -S(0)dRa, -S(0)d NRaRb, -(CH2)nS(0)dRa, - P(=0)RaRb, -C(=Y)Ra, -C(=Y)ORa, -C(=Y)SRa or -C(=Y)NRaRb, each of which may optionally be substituted at any available position by one or more substituents selected from Cr^alkyl, C2.i2alkenyl, C2-i2 alkynyl, C3-20cycloalkyl, heterocyclyl, aryl, heteroaryl, , -CN, -N02 or NH2;
Ra, Rb and Rc are independently selected from hydrogen, halogen, Ci_i2alkyl, C2-i2alkenyl, C2-i2alkynyl, C|-i2alkoxy, Ci-6alkoxyCi.6alkyl, C(.i2alkylcarbonyl, Ci_i2alkoxycarbonyl, C3-2ocycloalkyl, heterocyclyl, aryl, heteroaryl, -(CH2)n-cycloalkyl, -(CH2)n-heterocyclyl, -(CH2)n-aryl, -(CH2)n-heteroaryl, -CN, -N02, -N3, -NCS, -NR8R9, -(CH2)nNR8R9, - (CH2)nN3, -(CH2)„NCS, -CR8(=NOR9), -OH, -OR8, -CH2OH, -(CH2)nYR8 , - (CH2)nS(0)dR8, -(CH2)nS(0)dNR8R9, -(CH2)nP(=0)R8R9, -C(=Y)R8, -C(=Y)OR8, - C(=Y)SR8, -C(=Y)NR8R9, -(CH2)nC(=Y)R8, -(CH2)nC(=Y)OR8, -(CH2)nC(=Y)NR8R9, - (CH2)n-C(=Y)SR8, -OC(=Y)R8, -OC(=Y)OR8, -OC(=Y)NR8R9, -OP(=0)R8R9, - (CH2)„OC(=Y)R8 , -(CH2)nOC(=Y)OR8, -(CH2)nOC(=Y)NR8R9, -(CH2)„OP(=0)R8R9, - (CH2)„NR8C(=Y)R9, -(CH2)nNR8C(=Y)OR9, -(CH2)nNR10C(=Y)NR8R9, (CH2)nNR8S(0)dR9 or -(CH2)nNHP(=0)R8R9; each of which may optionally be substituted at any available position by one or more substituents selected from Rn; wherein Ra and Rb can be joined together to form a monocyclic or polycyclic ring, which may further contain one or more heteroatoms selected from but not limited to O, S, SO, S02, NR16, PR15, oxo or P(=0)R15; the ring thus formed may further be substituted at any available position by R1 1 ; Re is selected from -cycloalkyl, -aryl, -heteroaryl, -YR , -C(=Y)R8, -C(=Y)OR8, - C(=Y)NR8R9, -C(=Y)SR8, -OC(=Y)R8 , -OC(=Y)OR8 or -OC(=Y)NR8R9 ; each of which may optionally be substituted at any available position by one or more substituents selected from R1 1;
n is 0, 1 , 2, 3, 4 or 5;
d is 1 or 2;
m is 1, 2, 3, 4 or 5.
2. The compound according to claim 1 having the Formula la,

Formula la
or its pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, wherein: R1, R2, R3, R4, R5, R6, R7, U, V, W, E, G, ring A and ring B are as defined in claim 1.
3. The compound according to claim 1 having the Formula lb,

Formula lb
or its pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers polymorphs, prodrugs, metabolites, salts or solvates thereof, wherein: R1, R2, R3, R4, R5 R6, R7, E, G, ring A and ring B are as defined in claim 1.
4. The compound according to claim 1 having the Formula Ic,

Formula Ic or its pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, wherein: R1, R2, R3, R4, R5, R°, R', E and G are as defined in claim 1.
5. The compound according to claim 1 having the Formula Id,

Formula Id
or its pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, wherein: R5, R6, R7, E and G are as defined in claim 1.
6. The compound according to claims 1 to 5, wherein E is selected from O or CH2.
7. The compound according to claims 1 to 5, wherein G is C1-12 alkylene which is unsubstituted or substituted at any available position by one or more substituents selected from R1 1.
8. The compound according to claims 1 to 5, wherein R7 is selected from the group consisting of -OR8, -(CH2)nYR8, -(CH2)„NR8R9, -(CH2)nNR10C(=Y)NR8R9, -
(CH2)nOC(=Y)R8 and -(CH2)nOC(=Y)OR8, each of which is unsubstituted or substituted, at any available position, with one or more substituents selected from R1 '.
9. The compound according to claims 1 to 5, wherein R7 is selected from the group consisting of -OCH3, -CH2OH, -CH2OCH2CF3, -CH2OCOCH3, -CH2OCOC2H5, - CH2OCOC3H7,
-CH2OCOC4H9, -CH2OCO(CH2)5CH3, -CH2OCO(CH2)7CH3, -CH2OCO(CH2)i0CH3, -CH2OCO(CH2)14CH3, -CH2OCOCH2OCOCH3, -CH2OCOOCH3, -CH2OCOOC2H5, - -CH2OCOOCH2C6H5, 
each of which is unsubstituted or substituted, at any available position, with one or more substituents selected from R1 1.
10. The compound according to claims 1 to 5, wherein R5 and R6 are independently selected from the group consisting of -H, -CH3, -S02CH3, -S02C2H5, -S02NH2, - S02N(CH3)2, -S02NHCOCH3, -S02-cycloalkyl, -S02-heterocyclyl, 
11. The compound according to clai gether with the N atom
to which they are attached represent  or
or
12. A compound which is selected from the group consisting of:






13. A pharmaceutical composition, comprising a compound according to claims 1 , 12 or its pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, optionally in combination with one or more pharmaceutically acceptable carrier(s).
14. A method for prophylaxis, amelioration and/or treatment of one or more conditions mediated by SGLT-2, in a subject in need thereof, which comprises administering a therapeutically effective amount of compound according to claims 1, 12 or its pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
15. A method for the prophylaxis, amelioration and/or treatment of one or more diseases, disorders and conditions selected from the group consisting of diabetes (including Type I and Type II), Metabolic Syndrome or 'Syndrome X' including impaired glucose tolerance, insulin resistance, metabolic acidosis or ketosis, disorders of food intake, satiety disorders, obesity, hyperinsulinemia, dyslipidemia (including hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels, high LDL levels), hypertension associated with metabolic disorders, congestive heart failure, edema, hyperuricemia, gout, wound healing, tissue '•ischemia, which comprises administering a therapeutically effective amount of compound according to claims 1 , 12 or its pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
16. A method for the prophylaxis, amelioration and/or treatment of the diseases, disorders and conditions collectively referenced to as "diabetic complications" which include both acute complications and chronic complication, which comprises administering a therapeutically effective amount of compound according to claims 1, 12 or its pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof.
17. Use of a compound according to claims 1 , 12 or its pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, for the manufacture of a medicament for the prophylaxis, amelioration and/or treatment of one or more conditions mediated by SGLT-2, in a subject in need thereof.
18. Use of a compound according to claims 1 , 12 or its pharmaceutically acceptable derivatives, analogs, tautomeric forms, isomers, polymorphs, prodrugs, metabolites, salts or solvates thereof, in combination with other therapeutic agents.
19. Use according to claim 17, wherein the medicament is administered orally, parenterally or topically.
20. The compounds of Formula I, methods and compositions as described and illustrated herein.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN2553/DEL/2009 | 2009-12-09 | ||
| IN2553DE2009 | 2009-12-09 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2011070592A2 true WO2011070592A2 (en) | 2011-06-16 |
| WO2011070592A3 WO2011070592A3 (en) | 2012-05-10 |
Family
ID=44145994
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2010/000796 WO2011070592A2 (en) | 2009-12-09 | 2010-12-09 | Novel sugar derivatives |
Country Status (2)
| Country | Link |
|---|---|
| AR (1) | AR079438A1 (en) |
| WO (1) | WO2011070592A2 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013038429A2 (en) | 2011-09-13 | 2013-03-21 | Panacea Biotec Ltd. | Novel sglt inhibitors |
| US8697658B2 (en) | 2011-12-15 | 2014-04-15 | National Health Research Institutes | Glycoside compounds |
| WO2014081660A1 (en) * | 2012-11-20 | 2014-05-30 | Lexicon Pharmaceuticals, Inc. | Inhibitors of sodium glucose cotransporter 1 |
| US9085517B2 (en) | 2011-12-15 | 2015-07-21 | Pfizer Limited | Sulfonamide derivatives |
| WO2021018044A1 (en) * | 2019-07-26 | 2021-02-04 | 南京明德新药研发有限公司 | Sglt2/dpp4 inhibitor and application thereof |
| WO2022160737A1 (en) * | 2021-01-26 | 2022-08-04 | 东宝紫星(杭州)生物医药有限公司 | Crystal form of tetrahydropyran ring compound and preparation method therefor |
Citations (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002064606A1 (en) | 2001-02-14 | 2002-08-22 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives and medicinal use thereof |
| WO2002088157A1 (en) | 2001-04-27 | 2002-11-07 | Ajinomoto Co., Inc. | N-substituted pyrazolyl-o-glycoside derivatives and remedial agent for diabetes containing the same |
| WO2003000712A1 (en) | 2001-06-20 | 2003-01-03 | Kissei Pharmaceutical Co., Ltd. | Nitrogenous heterocyclic derivative, medicinal composition containing the same, medicinal use thereof, and intermediate therefor |
| US6515117B2 (en) | 1999-10-12 | 2003-02-04 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| WO2003020737A1 (en) | 2001-09-05 | 2003-03-13 | Bristol-Myers Squibb Company | O-pyrazole glucoside sglt2 inhibitors and method of use |
| US6555519B2 (en) | 2000-03-30 | 2003-04-29 | Bristol-Myers Squibb Company | O-glucosylated benzamide SGLT2 inhibitors and method |
| US6683056B2 (en) | 2000-03-30 | 2004-01-27 | Bristol-Myers Squibb Company | O-aryl glucoside SGLT2 inhibitors and method |
| WO2004013118A1 (en) | 2002-08-05 | 2004-02-12 | Yamanouchi Pharmaceutical Co., Ltd. | Azulene derivatives and salts thereof |
| WO2004058790A1 (en) | 2002-12-25 | 2004-07-15 | Kissei Pharmaceutical Co., Ltd. | Nitrogen-containing heterocycic derivatives, medicinal compositions containing the same and medicinal use thereof |
| WO2004087727A1 (en) | 2003-03-31 | 2004-10-14 | Kissei Pharmaceutical Co., Ltd. | Fused heterocyclic derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2004089966A1 (en) | 2003-04-01 | 2004-10-21 | Taisho Pharmaceutical Co., Ltd. | METHOD FOR SELECTIVE PREPARATION OF HETEROARYL 5-THIO- β-D-ALDOHEXOPYRANOSIDE |
| WO2004099230A1 (en) | 2003-05-12 | 2004-11-18 | Fujisawa Pharmaceutical Co., Ltd. | Monosaccharide compounds |
| JP2004359630A (en) | 2003-06-06 | 2004-12-24 | Yamanouchi Pharmaceut Co Ltd | Difluorodiphenylmethane derivative and its salt |
| WO2005021566A2 (en) | 2003-08-26 | 2005-03-10 | Boehringer Ingelheim International Gmbh | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| US6872706B2 (en) | 2000-09-29 | 2005-03-29 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| US6936590B2 (en) | 2001-03-13 | 2005-08-30 | Bristol Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| WO2005085267A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Nitrogenous fused-ring derivatives, medicinal compositions containing the derivatives, and use thereof as drugs |
| WO2005085265A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005095429A1 (en) | 2004-03-31 | 2005-10-13 | Kissei Pharmaceutical Co., Ltd. | Phenol derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2006002912A1 (en) | 2004-07-06 | 2006-01-12 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyls, medicaments containing said compounds, the use thereof, and methods for producing the same |
| WO2006008038A1 (en) | 2004-07-17 | 2006-01-26 | Boehringer Ingelheim International Gmbh | Methylidene-d-xylopyranosyl-substituted and oxo-d-xylopyranosyl-substituted phenyls, medicaments containing these compounds, their use and method for the production thereof |
| WO2006011469A1 (en) | 2004-07-26 | 2006-02-02 | Chugai Seiyaku Kabushiki Kaisha | Novel cyclohexane derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| WO2006010557A1 (en) | 2004-07-27 | 2006-02-02 | Boehringer Ingelheim International Gmbh | D-glucopyranosyl phenyl-substituted cyclene, medicaments containing these compounds, their use, and method for the production thereof |
| WO2006034489A2 (en) | 2004-09-23 | 2006-03-30 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors and method for their production |
| WO2006037537A2 (en) | 2004-10-01 | 2006-04-13 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyls, medicaments containing said compounds, use thereof, and method for the production thereof |
| WO2006054629A1 (en) | 2004-11-18 | 2006-05-26 | Kissei Pharmaceutical Co., Ltd. | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| US7056892B2 (en) | 1999-08-31 | 2006-06-06 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2006064033A2 (en) | 2004-12-16 | 2006-06-22 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2006089872A1 (en) | 2005-02-23 | 2006-08-31 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted ( (hetero)arylethynyl-benzyd-benzene derivatives and use thereof as sodium-dependent glucose cotransporter 2 (sglt2) inhibitors |
| US7129381B2 (en) | 2000-11-30 | 2006-10-31 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2007000445A1 (en) | 2005-06-29 | 2007-01-04 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20070049537A1 (en) | 2005-08-30 | 2007-03-01 | Matthias Eckhardt | Glucopyranosyl-substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US7189702B2 (en) | 2001-02-26 | 2007-03-13 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxypyrazole derivatives and medicinal use thereof |
| US7202350B2 (en) | 2003-03-14 | 2007-04-10 | Astellas Pharma Inc. | C-glycoside derivatives and salts thereof |
| US7247616B2 (en) | 2000-11-02 | 2007-07-24 | Ajinomoto Co., Inc. | Pyrazole derivatives and diabetic medicine containing them |
| WO2007093610A1 (en) | 2006-02-15 | 2007-08-23 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzonitrile derivatives, pharmaceutical compositions containing such compounds, their use and process for their manufacture |
| WO2007126117A1 (en) | 2006-05-02 | 2007-11-08 | Taisho Pharmaceutical Co., Ltd. | Phenyl 5-thioglucoside compound |
| US7294618B2 (en) | 2001-02-27 | 2007-11-13 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxypyrazole derivatives and medicinal use thereof |
| WO2007136116A2 (en) | 2006-05-19 | 2007-11-29 | Taisho Pharmaceutical Co., Ltd. | C-phenyl glycitol compound for the treatment of diabetes |
| WO2008013277A1 (en) | 2006-07-27 | 2008-01-31 | Chugai Seiyaku Kabushiki Kaisha | Fused ring spiroketal derivative and use thereof as drug for treating diabetes |
| WO2008013321A1 (en) | 2006-07-28 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | Novel sglt inhibitors |
| WO2008020011A1 (en) | 2006-08-15 | 2008-02-21 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted cyclopropylbenzene derivatives, pharmaceutical compositions containing such compounds, their use as sglt inhibitors and process for their manufacture |
| WO2008042688A2 (en) | 2006-09-29 | 2008-04-10 | Lexicon Pharmaceuticals, Inc. | Phlorizin analogs as inhibitors of sodium glucose co-transporter 2 |
| WO2008069327A1 (en) | 2006-12-04 | 2008-06-12 | Mitsubishi Tanabe Pharma Corporation | CRYSTALLINE FORM OF 1- (β-D-GLUCOPYRANOSYL) -4 -METHYL- 3- [5- (4 -FLUOROPHENYL) -2-THIENYLMETHYL] BENZENE HEMIHYDRATE |
| WO2008090570A1 (en) | 2007-01-25 | 2008-07-31 | Panacea Biotec Ltd | Novel antimicrobials |
| WO2008116195A2 (en) | 2007-03-22 | 2008-09-25 | Bristol-Myers Squibb | Compositions comprising an sglt2 ingibitor for treating obesity |
| WO2008122014A1 (en) | 2007-04-02 | 2008-10-09 | Theracos, Inc. | Benzylic glycoside derivatives and methods of use |
| WO2009026537A1 (en) | 2007-08-23 | 2009-02-26 | Theracos, Inc. | Benzylbenzene derivatives and methods of use |
| WO2009116090A2 (en) | 2008-03-18 | 2009-09-24 | Panacea Biotec Limited | Novel antimicrobials |
| WO2010022313A2 (en) | 2008-08-22 | 2010-02-25 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PH12000002657B1 (en) * | 1999-10-12 | 2006-02-21 | Bristol Myers Squibb Co | C-aryl glucoside SGLT2 inhibitors |
-
2010
- 2010-12-09 WO PCT/IN2010/000796 patent/WO2011070592A2/en active Application Filing
- 2010-12-09 AR ARP100104559A patent/AR079438A1/en unknown
Patent Citations (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7056892B2 (en) | 1999-08-31 | 2006-06-06 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| US6515117B2 (en) | 1999-10-12 | 2003-02-04 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| US6683056B2 (en) | 2000-03-30 | 2004-01-27 | Bristol-Myers Squibb Company | O-aryl glucoside SGLT2 inhibitors and method |
| US6555519B2 (en) | 2000-03-30 | 2003-04-29 | Bristol-Myers Squibb Company | O-glucosylated benzamide SGLT2 inhibitors and method |
| US6872706B2 (en) | 2000-09-29 | 2005-03-29 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| US7247616B2 (en) | 2000-11-02 | 2007-07-24 | Ajinomoto Co., Inc. | Pyrazole derivatives and diabetic medicine containing them |
| US7129381B2 (en) | 2000-11-30 | 2006-10-31 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2002064606A1 (en) | 2001-02-14 | 2002-08-22 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives and medicinal use thereof |
| US7189702B2 (en) | 2001-02-26 | 2007-03-13 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxypyrazole derivatives and medicinal use thereof |
| US7294618B2 (en) | 2001-02-27 | 2007-11-13 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxypyrazole derivatives and medicinal use thereof |
| US6936590B2 (en) | 2001-03-13 | 2005-08-30 | Bristol Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| WO2002088157A1 (en) | 2001-04-27 | 2002-11-07 | Ajinomoto Co., Inc. | N-substituted pyrazolyl-o-glycoside derivatives and remedial agent for diabetes containing the same |
| WO2003000712A1 (en) | 2001-06-20 | 2003-01-03 | Kissei Pharmaceutical Co., Ltd. | Nitrogenous heterocyclic derivative, medicinal composition containing the same, medicinal use thereof, and intermediate therefor |
| WO2003020737A1 (en) | 2001-09-05 | 2003-03-13 | Bristol-Myers Squibb Company | O-pyrazole glucoside sglt2 inhibitors and method of use |
| WO2004013118A1 (en) | 2002-08-05 | 2004-02-12 | Yamanouchi Pharmaceutical Co., Ltd. | Azulene derivatives and salts thereof |
| WO2004058790A1 (en) | 2002-12-25 | 2004-07-15 | Kissei Pharmaceutical Co., Ltd. | Nitrogen-containing heterocycic derivatives, medicinal compositions containing the same and medicinal use thereof |
| US7202350B2 (en) | 2003-03-14 | 2007-04-10 | Astellas Pharma Inc. | C-glycoside derivatives and salts thereof |
| WO2004087727A1 (en) | 2003-03-31 | 2004-10-14 | Kissei Pharmaceutical Co., Ltd. | Fused heterocyclic derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2004089966A1 (en) | 2003-04-01 | 2004-10-21 | Taisho Pharmaceutical Co., Ltd. | METHOD FOR SELECTIVE PREPARATION OF HETEROARYL 5-THIO- β-D-ALDOHEXOPYRANOSIDE |
| WO2004099230A1 (en) | 2003-05-12 | 2004-11-18 | Fujisawa Pharmaceutical Co., Ltd. | Monosaccharide compounds |
| JP2004359630A (en) | 2003-06-06 | 2004-12-24 | Yamanouchi Pharmaceut Co Ltd | Difluorodiphenylmethane derivative and its salt |
| WO2005021566A2 (en) | 2003-08-26 | 2005-03-10 | Boehringer Ingelheim International Gmbh | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| WO2005085267A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Nitrogenous fused-ring derivatives, medicinal compositions containing the derivatives, and use thereof as drugs |
| WO2005085265A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005095429A1 (en) | 2004-03-31 | 2005-10-13 | Kissei Pharmaceutical Co., Ltd. | Phenol derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2006002912A1 (en) | 2004-07-06 | 2006-01-12 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyls, medicaments containing said compounds, the use thereof, and methods for producing the same |
| WO2006008038A1 (en) | 2004-07-17 | 2006-01-26 | Boehringer Ingelheim International Gmbh | Methylidene-d-xylopyranosyl-substituted and oxo-d-xylopyranosyl-substituted phenyls, medicaments containing these compounds, their use and method for the production thereof |
| WO2006011469A1 (en) | 2004-07-26 | 2006-02-02 | Chugai Seiyaku Kabushiki Kaisha | Novel cyclohexane derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| WO2006010557A1 (en) | 2004-07-27 | 2006-02-02 | Boehringer Ingelheim International Gmbh | D-glucopyranosyl phenyl-substituted cyclene, medicaments containing these compounds, their use, and method for the production thereof |
| WO2006034489A2 (en) | 2004-09-23 | 2006-03-30 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors and method for their production |
| WO2006037537A2 (en) | 2004-10-01 | 2006-04-13 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyls, medicaments containing said compounds, use thereof, and method for the production thereof |
| WO2006054629A1 (en) | 2004-11-18 | 2006-05-26 | Kissei Pharmaceutical Co., Ltd. | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| WO2006064033A2 (en) | 2004-12-16 | 2006-06-22 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2006089872A1 (en) | 2005-02-23 | 2006-08-31 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted ( (hetero)arylethynyl-benzyd-benzene derivatives and use thereof as sodium-dependent glucose cotransporter 2 (sglt2) inhibitors |
| WO2007000445A1 (en) | 2005-06-29 | 2007-01-04 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20070049537A1 (en) | 2005-08-30 | 2007-03-01 | Matthias Eckhardt | Glucopyranosyl-substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2007093610A1 (en) | 2006-02-15 | 2007-08-23 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzonitrile derivatives, pharmaceutical compositions containing such compounds, their use and process for their manufacture |
| WO2007126117A1 (en) | 2006-05-02 | 2007-11-08 | Taisho Pharmaceutical Co., Ltd. | Phenyl 5-thioglucoside compound |
| WO2007136116A2 (en) | 2006-05-19 | 2007-11-29 | Taisho Pharmaceutical Co., Ltd. | C-phenyl glycitol compound for the treatment of diabetes |
| WO2008013277A1 (en) | 2006-07-27 | 2008-01-31 | Chugai Seiyaku Kabushiki Kaisha | Fused ring spiroketal derivative and use thereof as drug for treating diabetes |
| WO2008013321A1 (en) | 2006-07-28 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | Novel sglt inhibitors |
| WO2008020011A1 (en) | 2006-08-15 | 2008-02-21 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted cyclopropylbenzene derivatives, pharmaceutical compositions containing such compounds, their use as sglt inhibitors and process for their manufacture |
| WO2008042688A2 (en) | 2006-09-29 | 2008-04-10 | Lexicon Pharmaceuticals, Inc. | Phlorizin analogs as inhibitors of sodium glucose co-transporter 2 |
| WO2008069327A1 (en) | 2006-12-04 | 2008-06-12 | Mitsubishi Tanabe Pharma Corporation | CRYSTALLINE FORM OF 1- (β-D-GLUCOPYRANOSYL) -4 -METHYL- 3- [5- (4 -FLUOROPHENYL) -2-THIENYLMETHYL] BENZENE HEMIHYDRATE |
| WO2008090570A1 (en) | 2007-01-25 | 2008-07-31 | Panacea Biotec Ltd | Novel antimicrobials |
| WO2008116195A2 (en) | 2007-03-22 | 2008-09-25 | Bristol-Myers Squibb | Compositions comprising an sglt2 ingibitor for treating obesity |
| WO2008122014A1 (en) | 2007-04-02 | 2008-10-09 | Theracos, Inc. | Benzylic glycoside derivatives and methods of use |
| WO2009026537A1 (en) | 2007-08-23 | 2009-02-26 | Theracos, Inc. | Benzylbenzene derivatives and methods of use |
| WO2009116090A2 (en) | 2008-03-18 | 2009-09-24 | Panacea Biotec Limited | Novel antimicrobials |
| WO2010022313A2 (en) | 2008-08-22 | 2010-02-25 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
Non-Patent Citations (3)
| Title |
|---|
| "Targeted prodrug design to optimize drug delivery", AAPS PHARMASCI, vol. 2, no. 1, 2000, pages E6 |
| NUCLEOSIDE, NUCLOETIDES & NUCLEIC ACIDS, vol. 20, no. 4-7, 2001, pages 649 - 652 |
| PERRIN, D.D.; ARMAREGO, W.L.F.: "Purification of Laboratory Chemicals", 1988, PERGAMON PRESS |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013038429A3 (en) * | 2011-09-13 | 2013-11-28 | Panacea Biotec Ltd. | Novel sglt inhibitors |
| WO2013038429A2 (en) | 2011-09-13 | 2013-03-21 | Panacea Biotec Ltd. | Novel sglt inhibitors |
| US9018249B2 (en) | 2011-09-13 | 2015-04-28 | Panacea Biotec Limited | SGLT inhibitors |
| US9085517B2 (en) | 2011-12-15 | 2015-07-21 | Pfizer Limited | Sulfonamide derivatives |
| US8697658B2 (en) | 2011-12-15 | 2014-04-15 | National Health Research Institutes | Glycoside compounds |
| CN108440614A (en) * | 2012-11-20 | 2018-08-24 | 莱西肯医药有限公司 | The inhibitor of sodium glucose cotransporter 1 |
| KR102165224B1 (en) * | 2012-11-20 | 2020-10-13 | 렉시컨 파마슈티컬스 인코퍼레이티드 | Inhibitors of sodium glucose cotransporter 1 |
| US9200025B2 (en) | 2012-11-20 | 2015-12-01 | Lexicon Pharmaceuticals, Inc. | Inhibitors of sodium glucose cotransporter 1 |
| JP2016504285A (en) * | 2012-11-20 | 2016-02-12 | レクシコン ファーマシューティカルズ インコーポレイテッド | Inhibitor of sodium glucose cotransporter 1 |
| US9688710B2 (en) | 2012-11-20 | 2017-06-27 | Lexicon Pharmaceuticals, Inc. | Inhibitors of sodium glucose cotransporter 1 |
| WO2014081660A1 (en) * | 2012-11-20 | 2014-05-30 | Lexicon Pharmaceuticals, Inc. | Inhibitors of sodium glucose cotransporter 1 |
| EP3489226A1 (en) * | 2012-11-20 | 2019-05-29 | Lexicon Pharmaceuticals, Inc. | Inhibitors of sodium glucose cotransporter 1 |
| KR20150085067A (en) * | 2012-11-20 | 2015-07-22 | 렉시컨 파마슈티컬스 인코퍼레이티드 | Inhibitors of sodium glucose cotransporter 1 |
| WO2021018044A1 (en) * | 2019-07-26 | 2021-02-04 | 南京明德新药研发有限公司 | Sglt2/dpp4 inhibitor and application thereof |
| CN114026079A (en) * | 2019-07-26 | 2022-02-08 | 南京明德新药研发有限公司 | SGLT2/DPP4 inhibitor and application thereof |
| JP2022533475A (en) * | 2019-07-26 | 2022-07-22 | メッドシャイン ディスカバリー インコーポレイテッド | SGLT2/DPP4 inhibitors and uses thereof |
| CN114026079B (en) * | 2019-07-26 | 2022-10-18 | 南京明德新药研发有限公司 | SGLT2/DPP4 inhibitor and application thereof |
| JP7227427B2 (en) | 2019-07-26 | 2023-02-21 | メッドシャイン ディスカバリー インコーポレイテッド | SGLT2/DPP4 inhibitors and uses thereof |
| AU2020320890B2 (en) * | 2019-07-26 | 2023-03-09 | Medshine Discovery Inc. | SGLT2/DPP4 inhibitor and application thereof |
| WO2022160737A1 (en) * | 2021-01-26 | 2022-08-04 | 东宝紫星(杭州)生物医药有限公司 | Crystal form of tetrahydropyran ring compound and preparation method therefor |
Also Published As
| Publication number | Publication date |
|---|---|
| AR079438A1 (en) | 2012-01-25 |
| WO2011070592A3 (en) | 2012-05-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2755722B1 (en) | Novel sglt inhibitors | |
| KR101837488B1 (en) | Optically pure benzyl-4-chlorophenyl-c-glucoside derivative | |
| EP2717689A2 (en) | Novel sglt inhibitors | |
| CN102159206A (en) | Glucoside derivatives and uses thereof as sglt inhibitors | |
| JP5908973B2 (en) | Glycoside derivatives and their use for the treatment of diabetes | |
| BR112012009053B1 (en) | GLYCOSIDE DERIVATIVES, THEIR USES, AND PHARMACEUTICAL COMPOSITIONS | |
| US20120196813A1 (en) | Glycoside derivative and uses thereof | |
| BRPI0717156B1 (en) | sodium glucose co-transporter 2 inhibitors and pharmaceutical composition comprising them | |
| WO2011070592A2 (en) | Novel sugar derivatives | |
| EP2099791A1 (en) | Thienyl-containing glycopyranosyl derivatives as antidiabetics | |
| WO2010018435A1 (en) | Amide glycosides | |
| RU2678327C2 (en) | Mannose derivatives for treating bacterial infections | |
| MX2011002989A (en) | Glycoside derivatives and uses thereof. | |
| US9828366B2 (en) | Bicyclic derivatives and pharmaceutical composition including the same | |
| KR20150130177A (en) | 2,3-dihydrobenzofuran derivatives as an sglt inhibitor and pharmaceutical composition comprising same | |
| CN114599643B (en) | Aryl glucoside derivative | |
| CN115003661A (en) | Aryl glucoside derivatives and their use in medicine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10807393 Country of ref document: EP Kind code of ref document: A2 |