WO2007129668A1 - Pyrazolyl 5-thioglucoside compound - Google Patents
Pyrazolyl 5-thioglucoside compound Download PDFInfo
- Publication number
- WO2007129668A1 WO2007129668A1 PCT/JP2007/059386 JP2007059386W WO2007129668A1 WO 2007129668 A1 WO2007129668 A1 WO 2007129668A1 JP 2007059386 W JP2007059386 W JP 2007059386W WO 2007129668 A1 WO2007129668 A1 WO 2007129668A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- pyrazolyl
- pharmaceutically acceptable
- hydrate
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 151
- 125000003226 pyrazolyl group Chemical group 0.000 title claims abstract description 29
- -1 pyrazolyl 5-thio-β-D-glucopyranoside compound Chemical class 0.000 claims abstract description 64
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 49
- 150000003839 salts Chemical class 0.000 claims abstract description 39
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 34
- 239000000126 substance Substances 0.000 claims abstract description 25
- 230000000694 effects Effects 0.000 claims abstract description 11
- 125000000217 alkyl group Chemical group 0.000 claims description 66
- 239000003814 drug Substances 0.000 claims description 24
- 239000003112 inhibitor Substances 0.000 claims description 20
- 125000002947 alkylene group Chemical group 0.000 claims description 18
- 229910052757 nitrogen Inorganic materials 0.000 claims description 16
- 125000001424 substituent group Chemical group 0.000 claims description 16
- 125000005037 alkyl phenyl group Chemical group 0.000 claims description 10
- 125000003545 alkoxy group Chemical group 0.000 claims description 7
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 125000004076 pyridyl group Chemical group 0.000 claims description 7
- 239000004480 active ingredient Substances 0.000 claims description 6
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 6
- 229940124597 therapeutic agent Drugs 0.000 claims description 6
- 125000004429 atom Chemical group 0.000 claims description 5
- 125000004432 carbon atom Chemical group C* 0.000 claims description 5
- 102000000070 Sodium-Glucose Transport Proteins Human genes 0.000 claims description 4
- 108010080361 Sodium-Glucose Transport Proteins Proteins 0.000 claims description 4
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 4
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 4
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- 125000004434 sulfur atom Chemical group 0.000 claims description 3
- 125000005842 heteroatom Chemical group 0.000 claims description 2
- 229910052727 yttrium Inorganic materials 0.000 claims description 2
- 125000004450 alkenylene group Chemical group 0.000 claims 1
- 230000000069 prophylactic effect Effects 0.000 claims 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 abstract description 15
- 239000008103 glucose Substances 0.000 abstract description 15
- 208000002705 Glucose Intolerance Diseases 0.000 abstract description 14
- 201000009104 prediabetes syndrome Diseases 0.000 abstract description 14
- 238000010521 absorption reaction Methods 0.000 abstract description 6
- 125000005843 halogen group Chemical group 0.000 abstract description 4
- 210000001035 gastrointestinal tract Anatomy 0.000 abstract description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 abstract 2
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 abstract 1
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 abstract 1
- 125000006590 (C2-C6) alkenylene group Chemical group 0.000 abstract 1
- 108091006277 SLC5A1 Proteins 0.000 abstract 1
- 102000058090 Sodium-Glucose Transporter 1 Human genes 0.000 abstract 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 84
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 81
- 238000006243 chemical reaction Methods 0.000 description 42
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 39
- 239000000243 solution Substances 0.000 description 36
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 33
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical class CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 31
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 30
- 238000004519 manufacturing process Methods 0.000 description 27
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 26
- 239000002904 solvent Substances 0.000 description 26
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 25
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 24
- 239000000203 mixture Substances 0.000 description 22
- 238000000034 method Methods 0.000 description 20
- 238000010898 silica gel chromatography Methods 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 18
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 18
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 18
- 206010012601 diabetes mellitus Diseases 0.000 description 18
- 230000002829 reductive effect Effects 0.000 description 17
- 229940079593 drug Drugs 0.000 description 16
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 16
- 238000005481 NMR spectroscopy Methods 0.000 description 15
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 15
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 15
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 15
- 238000002360 preparation method Methods 0.000 description 15
- 238000003786 synthesis reaction Methods 0.000 description 15
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 14
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 14
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 14
- 125000006179 2-methyl benzyl group Chemical group [H]C1=C([H])C(=C(C([H])=C1[H])C([H])([H])*)C([H])([H])[H] 0.000 description 13
- 239000002585 base Substances 0.000 description 13
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 13
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 13
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 12
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 12
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 11
- 239000008280 blood Substances 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 11
- 239000000843 powder Substances 0.000 description 11
- 238000012360 testing method Methods 0.000 description 11
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 10
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 10
- 238000005160 1H NMR spectroscopy Methods 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 9
- 150000003217 pyrazoles Chemical class 0.000 description 9
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 8
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 8
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 8
- 229910000024 caesium carbonate Inorganic materials 0.000 description 8
- 229910052763 palladium Inorganic materials 0.000 description 8
- 229910000027 potassium carbonate Inorganic materials 0.000 description 8
- 239000011541 reaction mixture Substances 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 229940058020 2-amino-2-methyl-1-propanol Drugs 0.000 description 7
- 241000700159 Rattus Species 0.000 description 7
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 7
- CBTVGIZVANVGBH-UHFFFAOYSA-N aminomethyl propanol Chemical compound CC(C)(N)CO CBTVGIZVANVGBH-UHFFFAOYSA-N 0.000 description 7
- 239000003054 catalyst Substances 0.000 description 7
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 7
- 230000002401 inhibitory effect Effects 0.000 description 7
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 7
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 7
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- 101000716688 Homo sapiens Sodium/glucose cotransporter 1 Proteins 0.000 description 6
- 101000716682 Homo sapiens Sodium/glucose cotransporter 2 Proteins 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- ABBZJHFBQXYTLU-UHFFFAOYSA-N but-3-enamide Chemical compound NC(=O)CC=C ABBZJHFBQXYTLU-UHFFFAOYSA-N 0.000 description 6
- 229910000019 calcium carbonate Inorganic materials 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 102000052194 human SLC5A1 Human genes 0.000 description 6
- 102000052543 human SLC5A2 Human genes 0.000 description 6
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 6
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 230000009102 absorption Effects 0.000 description 5
- 229960000583 acetic acid Drugs 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 238000010511 deprotection reaction Methods 0.000 description 5
- 239000002274 desiccant Substances 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 229910052739 hydrogen Inorganic materials 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 235000019341 magnesium sulphate Nutrition 0.000 description 5
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 5
- 210000000813 small intestine Anatomy 0.000 description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- SCVJRXQHFJXZFZ-KVQBGUIXSA-N 2-amino-9-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-3h-purine-6-thione Chemical compound C1=2NC(N)=NC(=S)C=2N=CN1[C@H]1C[C@H](O)[C@@H](CO)O1 SCVJRXQHFJXZFZ-KVQBGUIXSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- 208000002249 Diabetes Complications Diseases 0.000 description 4
- 206010012655 Diabetic complications Diseases 0.000 description 4
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- 238000006751 Mitsunobu reaction Methods 0.000 description 4
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 4
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 4
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- DNSISZSEWVHGLH-UHFFFAOYSA-N butanamide Chemical compound CCCC(N)=O DNSISZSEWVHGLH-UHFFFAOYSA-N 0.000 description 4
- 239000004202 carbamide Substances 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 4
- 201000010099 disease Diseases 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- PWWSSIYVTQUJQQ-UHFFFAOYSA-N distearyl thiodipropionate Chemical compound CCCCCCCCCCCCCCCCCCOC(=O)CCSCCC(=O)OCCCCCCCCCCCCCCCCCC PWWSSIYVTQUJQQ-UHFFFAOYSA-N 0.000 description 4
- 230000004190 glucose uptake Effects 0.000 description 4
- 229940125396 insulin Drugs 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 4
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 4
- UGRMITBWUVWUEB-UHFFFAOYSA-N 1-$l^{1}-oxidanyl-3-methylbenzene Chemical group CC1=CC=CC([O])=C1 UGRMITBWUVWUEB-UHFFFAOYSA-N 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 3
- WRLSNQXIGKWPGJ-UHFFFAOYSA-N 2-methylpropanamide Chemical group C[C](C)C(N)=O WRLSNQXIGKWPGJ-UHFFFAOYSA-N 0.000 description 3
- 125000006281 4-bromobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1Br)C([H])([H])* 0.000 description 3
- 206010002091 Anaesthesia Diseases 0.000 description 3
- 108010011459 Exenatide Proteins 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- 102000004877 Insulin Human genes 0.000 description 3
- 108090001061 Insulin Proteins 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 3
- 102000003728 Peroxisome Proliferator-Activated Receptors Human genes 0.000 description 3
- 108090000029 Peroxisome Proliferator-Activated Receptors Proteins 0.000 description 3
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 102100037202 Sodium/myo-inositol cotransporter 2 Human genes 0.000 description 3
- 101710090560 Sodium/myo-inositol cotransporter 2 Proteins 0.000 description 3
- 239000007983 Tris buffer Substances 0.000 description 3
- 239000000556 agonist Substances 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 230000037005 anaesthesia Effects 0.000 description 3
- 239000000883 anti-obesity agent Substances 0.000 description 3
- 229940125710 antiobesity agent Drugs 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 3
- PVEOYINWKBTPIZ-UHFFFAOYSA-N but-3-enoic acid Chemical compound OC(=O)CC=C PVEOYINWKBTPIZ-UHFFFAOYSA-N 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 238000006264 debenzylation reaction Methods 0.000 description 3
- 235000005911 diet Nutrition 0.000 description 3
- 238000001647 drug administration Methods 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 125000001841 imino group Chemical group [H]N=* 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- LFQULJPVXNYWAG-UHFFFAOYSA-N sodium;phenylmethanolate Chemical compound [Na]OCC1=CC=CC=C1 LFQULJPVXNYWAG-UHFFFAOYSA-N 0.000 description 3
- 210000000952 spleen Anatomy 0.000 description 3
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 3
- 230000007704 transition Effects 0.000 description 3
- IJJLRUSZMLMXCN-SLPGGIOYSA-N (2r,3r,4s,5r)-2,3,4,6-tetrahydroxy-5-sulfanylhexanal Chemical class OC[C@@H](S)[C@@H](O)[C@H](O)[C@@H](O)C=O IJJLRUSZMLMXCN-SLPGGIOYSA-N 0.000 description 2
- YCWRFIYBUQBHJI-UHFFFAOYSA-N 2-(4-aminophenyl)acetonitrile Chemical group NC1=CC=C(CC#N)C=C1 YCWRFIYBUQBHJI-UHFFFAOYSA-N 0.000 description 2
- FHEYFIGWYQJVDR-ACJLOTCBSA-N 2-[[3-[(2r)-2-[[(2r)-2-(3-chlorophenyl)-2-hydroxyethyl]amino]propyl]-1h-indol-7-yl]oxy]acetic acid Chemical compound C1([C@@H](O)CN[C@@H](CC=2C3=CC=CC(OCC(O)=O)=C3NC=2)C)=CC=CC(Cl)=C1 FHEYFIGWYQJVDR-ACJLOTCBSA-N 0.000 description 2
- 239000001763 2-hydroxyethyl(trimethyl)azanium Substances 0.000 description 2
- WZFZJEPHYDDFCT-UHFFFAOYSA-N 3-chloro-2-methyl-n-[4-[2-(3-oxomorpholin-4-yl)ethyl]-1,3-thiazol-2-yl]benzenesulfonamide Chemical compound CC1=C(Cl)C=CC=C1S(=O)(=O)NC1=NC(CCN2C(COCC2)=O)=CS1 WZFZJEPHYDDFCT-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- JBMKAUGHUNFTOL-UHFFFAOYSA-N Aldoclor Chemical class C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NC=NS2(=O)=O JBMKAUGHUNFTOL-UHFFFAOYSA-N 0.000 description 2
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- 235000019743 Choline chloride Nutrition 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 229920000858 Cyclodextrin Polymers 0.000 description 2
- 101710088194 Dehydrogenase Proteins 0.000 description 2
- HTQBXNHDCUEHJF-XWLPCZSASA-N Exenatide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 HTQBXNHDCUEHJF-XWLPCZSASA-N 0.000 description 2
- 101710198884 GATA-type zinc finger protein 1 Proteins 0.000 description 2
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 2
- 238000007341 Heck reaction Methods 0.000 description 2
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 2
- 206010020772 Hypertension Diseases 0.000 description 2
- YSDQQAXHVYUZIW-QCIJIYAXSA-N Liraglutide Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCNC(=O)CC[C@H](NC(=O)CCCCCCCCCCCCCCC)C(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=C(O)C=C1 YSDQQAXHVYUZIW-QCIJIYAXSA-N 0.000 description 2
- 108010019598 Liraglutide Proteins 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- OKJHGOPITGTTIM-DEOSSOPVSA-N Naveglitazar Chemical compound C1=CC(C[C@H](OC)C(O)=O)=CC=C1OCCCOC(C=C1)=CC=C1OC1=CC=CC=C1 OKJHGOPITGTTIM-DEOSSOPVSA-N 0.000 description 2
- 108010025020 Nerve Growth Factor Proteins 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 102100040918 Pro-glucagon Human genes 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 229940123518 Sodium/glucose cotransporter 2 inhibitor Drugs 0.000 description 2
- ZSJLQEPLLKMAKR-UHFFFAOYSA-N Streptozotocin Natural products O=NN(C)C(=O)NC1C(O)OC(CO)C(O)C1O ZSJLQEPLLKMAKR-UHFFFAOYSA-N 0.000 description 2
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 2
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 150000001336 alkenes Chemical class 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 239000003146 anticoagulant agent Substances 0.000 description 2
- 229940030600 antihypertensive agent Drugs 0.000 description 2
- 239000002220 antihypertensive agent Substances 0.000 description 2
- 239000003524 antilipemic agent Substances 0.000 description 2
- 229960004676 antithrombotic agent Drugs 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 159000000007 calcium salts Chemical class 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- SGMZJAMFUVOLNK-UHFFFAOYSA-M choline chloride Chemical compound [Cl-].C[N+](C)(C)CCO SGMZJAMFUVOLNK-UHFFFAOYSA-M 0.000 description 2
- 229960003178 choline chloride Drugs 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 230000037213 diet Effects 0.000 description 2
- 239000002934 diuretic Substances 0.000 description 2
- 229940030606 diuretics Drugs 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 229960001519 exenatide Drugs 0.000 description 2
- 239000013604 expression vector Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 229920000669 heparin Polymers 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 210000004347 intestinal mucosa Anatomy 0.000 description 2
- 229960001021 lactose monohydrate Drugs 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- 239000012046 mixed solvent Substances 0.000 description 2
- 229960000698 nateglinide Drugs 0.000 description 2
- OELFLUMRDSZNSF-BRWVUGGUSA-N nateglinide Chemical compound C1C[C@@H](C(C)C)CC[C@@H]1C(=O)N[C@@H](C(O)=O)CC1=CC=CC=C1 OELFLUMRDSZNSF-BRWVUGGUSA-N 0.000 description 2
- 210000005036 nerve Anatomy 0.000 description 2
- 235000001968 nicotinic acid Nutrition 0.000 description 2
- 239000011664 nicotinic acid Substances 0.000 description 2
- QWVGKYWNOKOFNN-UHFFFAOYSA-N o-cresol Chemical compound CC1=CC=CC=C1O QWVGKYWNOKOFNN-UHFFFAOYSA-N 0.000 description 2
- 238000007410 oral glucose tolerance test Methods 0.000 description 2
- AHLBNYSZXLDEJQ-FWEHEUNISA-N orlistat Chemical compound CCCCCCCCCCC[C@H](OC(=O)[C@H](CC(C)C)NC=O)C[C@@H]1OC(=O)[C@H]1CCCCCC AHLBNYSZXLDEJQ-FWEHEUNISA-N 0.000 description 2
- 229960001243 orlistat Drugs 0.000 description 2
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 2
- DHHVAGZRUROJKS-UHFFFAOYSA-N phentermine Chemical compound CC(C)(N)CC1=CC=CC=C1 DHHVAGZRUROJKS-UHFFFAOYSA-N 0.000 description 2
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 2
- 239000002504 physiological saline solution Substances 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229910003446 platinum oxide Inorganic materials 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 230000003449 preventive effect Effects 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- NHZMQXZHNVQTQA-UHFFFAOYSA-N pyridoxamine Chemical compound CC1=NC=C(CO)C(CN)=C1O NHZMQXZHNVQTQA-UHFFFAOYSA-N 0.000 description 2
- 239000002464 receptor antagonist Substances 0.000 description 2
- 229940044551 receptor antagonist Drugs 0.000 description 2
- 159000000000 sodium salts Chemical class 0.000 description 2
- 229960001052 streptozocin Drugs 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- XOAAWQZATWQOTB-UHFFFAOYSA-N taurine Chemical compound NCCS(O)(=O)=O XOAAWQZATWQOTB-UHFFFAOYSA-N 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- 239000003451 thiazide diuretic agent Substances 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- DBGIVFWFUFKIQN-VIFPVBQESA-N (+)-Fenfluramine Chemical compound CCN[C@@H](C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-VIFPVBQESA-N 0.000 description 1
- DBGIVFWFUFKIQN-UHFFFAOYSA-N (+-)-Fenfluramine Chemical compound CCNC(C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-UHFFFAOYSA-N 0.000 description 1
- 108010000230 (1-(((1-(1-oxo-2-N-((phenylmethoxy)carbonyl)-3-phenylpropyl)pyrrolidin-2-yl)carbonyl)amino)-4-methoxybutyl)-1-boronic acid Proteins 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- VLPIATFUUWWMKC-SNVBAGLBSA-N (2r)-1-(2,6-dimethylphenoxy)propan-2-amine Chemical compound C[C@@H](N)COC1=C(C)C=CC=C1C VLPIATFUUWWMKC-SNVBAGLBSA-N 0.000 description 1
- XSNMGLZVFNDDPW-ZWKOTPCHSA-N (2s)-1-[(2r)-2-[3-chloro-5-(difluoromethoxy)phenyl]-2-hydroxyacetyl]-n-[[4-(n'-methoxycarbamimidoyl)phenyl]methyl]azetidine-2-carboxamide Chemical compound C1=CC(C(/N)=N/OC)=CC=C1CNC(=O)[C@H]1N(C(=O)[C@H](O)C=2C=C(OC(F)F)C=C(Cl)C=2)CC1 XSNMGLZVFNDDPW-ZWKOTPCHSA-N 0.000 description 1
- ZXEIEKDGPVTZLD-NDEPHWFRSA-N (2s)-2-dodecylsulfanyl-n-(4-hydroxy-2,3,5-trimethylphenyl)-2-phenylacetamide Chemical compound O=C([C@@H](SCCCCCCCCCCCC)C=1C=CC=CC=1)NC1=CC(C)=C(O)C(C)=C1C ZXEIEKDGPVTZLD-NDEPHWFRSA-N 0.000 description 1
- GYIYAOUGKJSCCG-HTRCEHHLSA-N (2s,3r)-2-[(6-aminopyridin-3-yl)methyl]-3-sulfanylbutanoic acid Chemical compound C[C@@H](S)[C@H](C(O)=O)CC1=CC=C(N)N=C1 GYIYAOUGKJSCCG-HTRCEHHLSA-N 0.000 description 1
- JSYGLDMGERSRPC-FQUUOJAGSA-N (2s,4s)-4-fluoro-1-[2-[[(1r,3s)-3-(1,2,4-triazol-1-ylmethyl)cyclopentyl]amino]acetyl]pyrrolidine-2-carbonitrile Chemical compound C1[C@@H](F)C[C@@H](C#N)N1C(=O)CN[C@H]1C[C@@H](CN2N=CN=C2)CC1 JSYGLDMGERSRPC-FQUUOJAGSA-N 0.000 description 1
- VMDKRSNUUUUARH-MQDBWYGVSA-N (3s)-4-[[(2s)-1-[[(2s)-2-[[(2s)-3-(1h-indol-3-yl)-2-[[2-[[(2s)-2-[[2-(4-sulfooxyphenyl)acetyl]amino]hexanoyl]amino]acetyl]amino]propanoyl]amino]hexanoyl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(methylamino)-4-oxobutanoic acid Chemical compound N([C@@H](CCCC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCCC)C(=O)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC(O)=O)NC)C(=O)CC1=CC=C(OS(O)(=O)=O)C=C1 VMDKRSNUUUUARH-MQDBWYGVSA-N 0.000 description 1
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 1
- VLSDXINSOMDCBK-BQYQJAHWSA-N (E)-1,1'-azobis(N,N-dimethylformamide) Chemical compound CN(C)C(=O)\N=N\C(=O)N(C)C VLSDXINSOMDCBK-BQYQJAHWSA-N 0.000 description 1
- KWTSXDURSIMDCE-QMMMGPOBSA-N (S)-amphetamine Chemical compound C[C@H](N)CC1=CC=CC=C1 KWTSXDURSIMDCE-QMMMGPOBSA-N 0.000 description 1
- BFUXUGOZJVHVMR-UHFFFAOYSA-N 1,1-dioxo-3,4-dihydro-2h-1$l^{6},2,4-benzothiadiazine-7-sulfonamide Chemical compound N1CNS(=O)(=O)C2=CC(S(=O)(=O)N)=CC=C21 BFUXUGOZJVHVMR-UHFFFAOYSA-N 0.000 description 1
- QWUWMCYKGHVNAV-UHFFFAOYSA-N 1,2-dihydrostilbene Chemical group C=1C=CC=CC=1CCC1=CC=CC=C1 QWUWMCYKGHVNAV-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- WTAPZWXVSZMMDG-UHFFFAOYSA-N 1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].C=1C=CC=CC=1C=CC(=O)C=CC1=CC=CC=C1 WTAPZWXVSZMMDG-UHFFFAOYSA-N 0.000 description 1
- KMXPHBJUGYLXDM-LSDHHAIUSA-N 1-[(7r,8s)-7-hydroxy-6,6-dimethyl-7,8-dihydropyrano[2,3-f][2,1,3]benzoxadiazol-8-yl]piperidin-2-one Chemical compound N1([C@H]2C3=CC4=NON=C4C=C3OC([C@@H]2O)(C)C)CCCCC1=O KMXPHBJUGYLXDM-LSDHHAIUSA-N 0.000 description 1
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 1
- 125000004825 2,2-dimethylpropylene group Chemical group [H]C([H])([H])C(C([H])([H])[H])(C([H])([H])[*:1])C([H])([H])[*:2] 0.000 description 1
- ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 2,3-dimethylbutane Chemical group CC(C)C(C)C ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 0.000 description 1
- NMRWDFUZLLQSBN-UHFFFAOYSA-N 2,4-dichloro-n-(3,5-dichloro-4-quinolin-3-yloxyphenyl)benzenesulfonamide Chemical compound ClC1=CC(Cl)=CC=C1S(=O)(=O)NC(C=C1Cl)=CC(Cl)=C1OC1=CN=C(C=CC=C2)C2=C1 NMRWDFUZLLQSBN-UHFFFAOYSA-N 0.000 description 1
- OFJRNBWSFXEHSA-UHFFFAOYSA-N 2-(3-amino-1,2-benzoxazol-5-yl)-n-[4-[2-[(dimethylamino)methyl]imidazol-1-yl]-2-fluorophenyl]-5-(trifluoromethyl)pyrazole-3-carboxamide Chemical compound CN(C)CC1=NC=CN1C(C=C1F)=CC=C1NC(=O)C1=CC(C(F)(F)F)=NN1C1=CC=C(ON=C2N)C2=C1 OFJRNBWSFXEHSA-UHFFFAOYSA-N 0.000 description 1
- DDTQLPXXNHLBAB-UHFFFAOYSA-N 2-(4-chlorophenyl)-2-[3-(trifluoromethyl)phenoxy]acetic acid Chemical compound C=1C=C(Cl)C=CC=1C(C(=O)O)OC1=CC=CC(C(F)(F)F)=C1 DDTQLPXXNHLBAB-UHFFFAOYSA-N 0.000 description 1
- 125000006516 2-(benzyloxy)ethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- SFRDXVJWXWOTEW-UHFFFAOYSA-N 2-(hydroxymethyl)propane-1,3-diol Chemical compound OCC(CO)CO SFRDXVJWXWOTEW-UHFFFAOYSA-N 0.000 description 1
- CMLUGNQVANVZHY-POURPWNDSA-N 2-[1-[2-[(3r,5s)-1-(3-acetyloxy-2,2-dimethylpropyl)-7-chloro-5-(2,3-dimethoxyphenyl)-2-oxo-5h-4,1-benzoxazepin-3-yl]acetyl]piperidin-4-yl]acetic acid Chemical compound COC1=CC=CC([C@@H]2C3=CC(Cl)=CC=C3N(CC(C)(C)COC(C)=O)C(=O)[C@@H](CC(=O)N3CCC(CC(O)=O)CC3)O2)=C1OC CMLUGNQVANVZHY-POURPWNDSA-N 0.000 description 1
- NSVFSAJIGAJDMR-UHFFFAOYSA-N 2-[benzyl(phenyl)amino]ethyl 5-(5,5-dimethyl-2-oxido-1,3,2-dioxaphosphinan-2-yl)-2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3-carboxylate Chemical compound CC=1NC(C)=C(C(=O)OCCN(CC=2C=CC=CC=2)C=2C=CC=CC=2)C(C=2C=C(C=CC=2)[N+]([O-])=O)C=1P1(=O)OCC(C)(C)CO1 NSVFSAJIGAJDMR-UHFFFAOYSA-N 0.000 description 1
- ZIOBGZDFMKCKGZ-UHFFFAOYSA-N 2-amino-2-methylpropanamide Chemical compound CC(C)(N)C(N)=O ZIOBGZDFMKCKGZ-UHFFFAOYSA-N 0.000 description 1
- UXFQFBNBSPQBJW-UHFFFAOYSA-N 2-amino-2-methylpropane-1,3-diol Chemical compound OCC(N)(C)CO UXFQFBNBSPQBJW-UHFFFAOYSA-N 0.000 description 1
- JIVPVXMEBJLZRO-CQSZACIVSA-N 2-chloro-5-[(1r)-1-hydroxy-3-oxo-2h-isoindol-1-yl]benzenesulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC([C@@]2(O)C3=CC=CC=C3C(=O)N2)=C1 JIVPVXMEBJLZRO-CQSZACIVSA-N 0.000 description 1
- JCCBZCMSYUSCFM-UHFFFAOYSA-N 2-chlorobenzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1Cl JCCBZCMSYUSCFM-UHFFFAOYSA-N 0.000 description 1
- ISPYQTSUDJAMAB-UHFFFAOYSA-N 2-chlorophenol Chemical compound OC1=CC=CC=C1Cl ISPYQTSUDJAMAB-UHFFFAOYSA-N 0.000 description 1
- YZQLWPMZQVHJED-UHFFFAOYSA-N 2-methylpropanethioic acid S-[2-[[[1-(2-ethylbutyl)cyclohexyl]-oxomethyl]amino]phenyl] ester Chemical compound C=1C=CC=C(SC(=O)C(C)C)C=1NC(=O)C1(CC(CC)CC)CCCCC1 YZQLWPMZQVHJED-UHFFFAOYSA-N 0.000 description 1
- MDBARDSTXONTFS-MNDUUMEHSA-N 3-[3-[3-methyl-4-[[5-propan-2-yl-3-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-1h-pyrazol-4-yl]methyl]phenoxy]propylamino]propanamide Chemical compound C=1C=C(OCCCNCCC(N)=O)C=C(C)C=1CC1=C(C(C)C)NN=C1O[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O MDBARDSTXONTFS-MNDUUMEHSA-N 0.000 description 1
- SRFCAWATPLCLMG-UHFFFAOYSA-N 3-[3-ethoxy-1-[[4-[(2-phenyl-1,3-thiazol-4-yl)methoxy]phenyl]methyl]pyrazol-4-yl]propanoic acid Chemical compound C1=C(CCC(O)=O)C(OCC)=NN1CC(C=C1)=CC=C1OCC1=CSC(C=2C=CC=CC=2)=N1 SRFCAWATPLCLMG-UHFFFAOYSA-N 0.000 description 1
- DGENZVKCTGIDRZ-UHFFFAOYSA-N 3-[4-[(3-phenoxyphenyl)methylamino]phenyl]propanoic acid Chemical compound C1=CC(CCC(=O)O)=CC=C1NCC1=CC=CC(OC=2C=CC=CC=2)=C1 DGENZVKCTGIDRZ-UHFFFAOYSA-N 0.000 description 1
- MCFBUIIRFZBRCU-UHFFFAOYSA-N 4-[1-[5-[6-(trifluoromethyl)-1h-benzimidazol-2-yl]pyridin-2-yl]piperidin-4-yl]oxycyclohexane-1-carboxylic acid Chemical compound C1CC(C(=O)O)CCC1OC1CCN(C=2N=CC(=CC=2)C=2NC3=CC(=CC=C3N=2)C(F)(F)F)CC1 MCFBUIIRFZBRCU-UHFFFAOYSA-N 0.000 description 1
- KJTRXVXWSSPHRV-UHFFFAOYSA-N 4-benzoyl-5-methyl-2-phenyl-1h-pyrazol-3-one Chemical compound O=C1C(C(=O)C=2C=CC=CC=2)=C(C)NN1C1=CC=CC=C1 KJTRXVXWSSPHRV-UHFFFAOYSA-N 0.000 description 1
- GPOQODYGMUTOQL-UHFFFAOYSA-N 4-bromo-3-methylphenol Chemical compound CC1=CC(O)=CC=C1Br GPOQODYGMUTOQL-UHFFFAOYSA-N 0.000 description 1
- GZFGOTFRPZRKDS-UHFFFAOYSA-N 4-bromophenol Chemical compound OC1=CC=C(Br)C=C1 GZFGOTFRPZRKDS-UHFFFAOYSA-N 0.000 description 1
- JRHNIQQUVJOPQC-AQNFWKISSA-N 4-methyl-7-[(2r,3r,4s,5s)-3,4,5-trihydroxythian-2-yl]oxychromen-2-one Chemical compound C1=CC=2C(C)=CC(=O)OC=2C=C1O[C@@H]1SC[C@@H](O)[C@H](O)[C@H]1O JRHNIQQUVJOPQC-AQNFWKISSA-N 0.000 description 1
- HMXDWDSNPRNUKI-UHFFFAOYSA-N 5-(4-bromophenyl)-1-(2,4-dichlorophenyl)-4-ethyl-N-(1-piperidinyl)-3-pyrazolecarboxamide Chemical compound CCC=1C(C(=O)NN2CCCCC2)=NN(C=2C(=CC(Cl)=CC=2)Cl)C=1C1=CC=C(Br)C=C1 HMXDWDSNPRNUKI-UHFFFAOYSA-N 0.000 description 1
- MPLLLQUZNJSVTK-UHFFFAOYSA-N 5-[3-[4-[2-(4-fluorophenyl)ethoxy]phenyl]propyl]furan-2-carboxylic acid Chemical compound O1C(C(=O)O)=CC=C1CCCC(C=C1)=CC=C1OCCC1=CC=C(F)C=C1 MPLLLQUZNJSVTK-UHFFFAOYSA-N 0.000 description 1
- AJJISMLYIMQAKP-OAHLLOKOSA-N 5-[4-[(2r)-4-(3-fluoro-4-methylsulfonylphenoxy)butan-2-yl]piperidin-1-yl]-3-propan-2-yl-1,2,4-oxadiazole Chemical compound CC(C)C1=NOC(N2CCC(CC2)[C@H](C)CCOC=2C=C(F)C(=CC=2)S(C)(=O)=O)=N1 AJJISMLYIMQAKP-OAHLLOKOSA-N 0.000 description 1
- BKYKPTRYDKTTJY-UHFFFAOYSA-N 6-chloro-3-(cyclopentylmethyl)-1,1-dioxo-3,4-dihydro-2H-1$l^{6},2,4-benzothiadiazine-7-sulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1NC2CC1CCCC1 BKYKPTRYDKTTJY-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical class O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 1
- 239000005541 ACE inhibitor Substances 0.000 description 1
- 108010093583 ART123 Proteins 0.000 description 1
- 102000011690 Adiponectin Human genes 0.000 description 1
- 108010076365 Adiponectin Proteins 0.000 description 1
- 229940118148 Aldose reductase inhibitor Drugs 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 229940123413 Angiotensin II antagonist Drugs 0.000 description 1
- QNZCBYKSOIHPEH-UHFFFAOYSA-N Apixaban Chemical compound C1=CC(OC)=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(CCCC3)=O)=C2C(C(N)=O)=N1 QNZCBYKSOIHPEH-UHFFFAOYSA-N 0.000 description 1
- 208000031104 Arterial Occlusive disease Diseases 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- PTQXTEKSNBVPQJ-UHFFFAOYSA-N Avasimibe Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1CC(=O)NS(=O)(=O)OC1=C(C(C)C)C=CC=C1C(C)C PTQXTEKSNBVPQJ-UHFFFAOYSA-N 0.000 description 1
- 108010028845 BIM 23190 Proteins 0.000 description 1
- 108700001281 BIM 51077 Proteins 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- XNCOSPRUTUOJCJ-UHFFFAOYSA-N Biguanide Chemical compound NC(N)=NC(N)=N XNCOSPRUTUOJCJ-UHFFFAOYSA-N 0.000 description 1
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 description 1
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- 239000002080 C09CA02 - Eprosartan Substances 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002081 C09CA05 - Tasosartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- GHOSNRCGJFBJIB-UHFFFAOYSA-N Candesartan cilexetil Chemical compound C=12N(CC=3C=CC(=CC=3)C=3C(=CC=CC=3)C3=NNN=N3)C(OCC)=NC2=CC=CC=1C(=O)OC(C)OC(=O)OC1CCCCC1 GHOSNRCGJFBJIB-UHFFFAOYSA-N 0.000 description 1
- 240000008574 Capsicum frutescens Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229940122072 Carbonic anhydrase inhibitor Drugs 0.000 description 1
- 102000003670 Carboxypeptidase B Human genes 0.000 description 1
- 108090000087 Carboxypeptidase B Proteins 0.000 description 1
- 102000003847 Carboxypeptidase B2 Human genes 0.000 description 1
- 108090000201 Carboxypeptidase B2 Proteins 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- RKWGIWYCVPQPMF-UHFFFAOYSA-N Chloropropamide Chemical compound CCCNC(=O)NS(=O)(=O)C1=CC=C(Cl)C=C1 RKWGIWYCVPQPMF-UHFFFAOYSA-N 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- BMOVQUBVGICXQN-UHFFFAOYSA-N Clinofibrate Chemical compound C1=CC(OC(C)(CC)C(O)=O)=CC=C1C1(C=2C=CC(OC(C)(CC)C(O)=O)=CC=2)CCCCC1 BMOVQUBVGICXQN-UHFFFAOYSA-N 0.000 description 1
- 206010010144 Completed suicide Diseases 0.000 description 1
- 102000034534 Cotransporters Human genes 0.000 description 1
- 108020003264 Cotransporters Proteins 0.000 description 1
- 229940127193 DGAT1 inhibitor Drugs 0.000 description 1
- JVHXJTBJCFBINQ-ADAARDCZSA-N Dapagliflozin Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)=CC=C1Cl JVHXJTBJCFBINQ-ADAARDCZSA-N 0.000 description 1
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 1
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 1
- 206010012689 Diabetic retinopathy Diseases 0.000 description 1
- DKMROQRQHGEIOW-UHFFFAOYSA-N Diethyl succinate Chemical compound CCOC(=O)CCC(=O)OCC DKMROQRQHGEIOW-UHFFFAOYSA-N 0.000 description 1
- 229940124213 Dipeptidyl peptidase 4 (DPP IV) inhibitor Drugs 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- VXLCNTLWWUDBSO-UHFFFAOYSA-N Ethiazide Chemical compound ClC1=C(S(N)(=O)=O)C=C2S(=O)(=O)NC(CC)NC2=C1 VXLCNTLWWUDBSO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 102100031562 Excitatory amino acid transporter 2 Human genes 0.000 description 1
- 102100023374 Forkhead box protein M1 Human genes 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 102100026148 Free fatty acid receptor 1 Human genes 0.000 description 1
- 102000027487 Fructose-Bisphosphatase Human genes 0.000 description 1
- 108010017464 Fructose-Bisphosphatase Proteins 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 101150116572 GLT-1 gene Proteins 0.000 description 1
- 229940100607 GPR119 agonist Drugs 0.000 description 1
- 229940125827 GPR40 agonist Drugs 0.000 description 1
- 206010017711 Gangrene Diseases 0.000 description 1
- 101800001586 Ghrelin Proteins 0.000 description 1
- 102400000442 Ghrelin-28 Human genes 0.000 description 1
- 102000051325 Glucagon Human genes 0.000 description 1
- 108060003199 Glucagon Proteins 0.000 description 1
- 229940089838 Glucagon-like peptide 1 receptor agonist Drugs 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- 229940121931 Gluconeogenesis inhibitor Drugs 0.000 description 1
- 206010018429 Glucose tolerance impaired Diseases 0.000 description 1
- 102000003638 Glucose-6-Phosphatase Human genes 0.000 description 1
- 108010086800 Glucose-6-Phosphatase Proteins 0.000 description 1
- 102000007390 Glycogen Phosphorylase Human genes 0.000 description 1
- 108010046163 Glycogen Phosphorylase Proteins 0.000 description 1
- 201000005569 Gout Diseases 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 101000907578 Homo sapiens Forkhead box protein M1 Proteins 0.000 description 1
- 101000912510 Homo sapiens Free fatty acid receptor 1 Proteins 0.000 description 1
- 101000976075 Homo sapiens Insulin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 102000004157 Hydrolases Human genes 0.000 description 1
- 108090000604 Hydrolases Proteins 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- GRRNUXAQVGOGFE-UHFFFAOYSA-N Hygromycin-B Natural products OC1C(NC)CC(N)C(O)C1OC1C2OC3(C(C(O)C(O)C(C(N)CO)O3)O)OC2C(O)C(CO)O1 GRRNUXAQVGOGFE-UHFFFAOYSA-N 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 206010060378 Hyperinsulinaemia Diseases 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- 201000001431 Hyperuricemia Diseases 0.000 description 1
- 239000003458 I kappa b kinase inhibitor Substances 0.000 description 1
- 229940126557 INCB13739 Drugs 0.000 description 1
- 206010022489 Insulin Resistance Diseases 0.000 description 1
- 229940122199 Insulin secretagogue Drugs 0.000 description 1
- 108010041872 Islet Amyloid Polypeptide Proteins 0.000 description 1
- 102000036770 Islet Amyloid Polypeptide Human genes 0.000 description 1
- KLDXJTOLSGUMSJ-JGWLITMVSA-N Isosorbide Chemical compound O[C@@H]1CO[C@@H]2[C@@H](O)CO[C@@H]21 KLDXJTOLSGUMSJ-JGWLITMVSA-N 0.000 description 1
- 206010023379 Ketoacidosis Diseases 0.000 description 1
- 208000007976 Ketosis Diseases 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 102000016267 Leptin Human genes 0.000 description 1
- 108010092277 Leptin Proteins 0.000 description 1
- 229940127470 Lipase Inhibitors Drugs 0.000 description 1
- 229940086609 Lipase inhibitor Drugs 0.000 description 1
- 239000012097 Lipofectamine 2000 Substances 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- ZPXSCAKFGYXMGA-UHFFFAOYSA-N Mazindol Chemical compound N12CCN=C2C2=CC=CC=C2C1(O)C1=CC=C(Cl)C=C1 ZPXSCAKFGYXMGA-UHFFFAOYSA-N 0.000 description 1
- 102000029828 Melanin-concentrating hormone receptor Human genes 0.000 description 1
- 108010047068 Melanin-concentrating hormone receptor Proteins 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- IBAQFPQHRJAVAV-ULAWRXDQSA-N Miglitol Chemical compound OCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO IBAQFPQHRJAVAV-ULAWRXDQSA-N 0.000 description 1
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- 108010045716 N-aminosulfonyldiphenylalanyl-prolyl-((4-amidinophenyl)methyl)amide Proteins 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- 102000015336 Nerve Growth Factor Human genes 0.000 description 1
- 102000007072 Nerve Growth Factors Human genes 0.000 description 1
- 102000003797 Neuropeptides Human genes 0.000 description 1
- 108090000189 Neuropeptides Proteins 0.000 description 1
- 108090000742 Neurotrophin 3 Proteins 0.000 description 1
- 102100029268 Neurotrophin-3 Human genes 0.000 description 1
- KUEUWHJGRZKESU-UHFFFAOYSA-N Niceritrol Chemical compound C=1C=CN=CC=1C(=O)OCC(COC(=O)C=1C=NC=CC=1)(COC(=O)C=1C=NC=CC=1)COC(=O)C1=CC=CN=C1 KUEUWHJGRZKESU-UHFFFAOYSA-N 0.000 description 1
- VRAHPESAMYMDQI-UHFFFAOYSA-N Nicomol Chemical compound C1CCC(COC(=O)C=2C=NC=CC=2)(COC(=O)C=2C=NC=CC=2)C(O)C1(COC(=O)C=1C=NC=CC=1)COC(=O)C1=CC=CN=C1 VRAHPESAMYMDQI-UHFFFAOYSA-N 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229940123973 Oxygen scavenger Drugs 0.000 description 1
- LXWSJMFOITXQMJ-UHFFFAOYSA-N P.CC1=C(C=CC=C1)O.CC1=C(C=CC=C1)O.CC1=C(C=CC=C1)O.C1(=CC=CC=C1)P(C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound P.CC1=C(C=CC=C1)O.CC1=C(C=CC=C1)O.CC1=C(C=CC=C1)O.C1(=CC=CC=C1)P(C1=CC=CC=C1)C1=CC=CC=C1 LXWSJMFOITXQMJ-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229940122907 Phosphatase inhibitor Drugs 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- CYLWJCABXYDINA-UHFFFAOYSA-N Polythiazide Polymers ClC1=C(S(N)(=O)=O)C=C2S(=O)(=O)N(C)C(CSCC(F)(F)F)NC2=C1 CYLWJCABXYDINA-UHFFFAOYSA-N 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 206010039509 Scab Diseases 0.000 description 1
- 241000555745 Sciuridae Species 0.000 description 1
- QLXKHBNJTPICNF-QMCAAQAGSA-N Sergliflozin etabonate Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](COC(=O)OCC)O[C@H]1OC1=CC=CC=C1CC1=CC=C(OC)C=C1 QLXKHBNJTPICNF-QMCAAQAGSA-N 0.000 description 1
- JLRNKCZRCMIVKA-UHFFFAOYSA-N Simfibrate Chemical compound C=1C=C(Cl)C=CC=1OC(C)(C)C(=O)OCCCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 JLRNKCZRCMIVKA-UHFFFAOYSA-N 0.000 description 1
- 101150041420 Slc1a2 gene Proteins 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- ABBQHOQBGMUPJH-UHFFFAOYSA-M Sodium salicylate Chemical compound [Na+].OC1=CC=CC=C1C([O-])=O ABBQHOQBGMUPJH-UHFFFAOYSA-M 0.000 description 1
- 102100033927 Sodium- and chloride-dependent GABA transporter 1 Human genes 0.000 description 1
- 101710104414 Sodium- and chloride-dependent GABA transporter 1 Proteins 0.000 description 1
- 229940127504 Somatostatin Receptor Agonists Drugs 0.000 description 1
- 229940123051 Somatostatin receptor agonist Drugs 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- FNYLWPVRPXGIIP-UHFFFAOYSA-N Triamterene Chemical compound NC1=NC2=NC(N)=NC(N)=C2N=C1C1=CC=CC=C1 FNYLWPVRPXGIIP-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- FZNCGRZWXLXZSZ-CIQUZCHMSA-N Voglibose Chemical compound OCC(CO)N[C@H]1C[C@](O)(CO)[C@@H](O)[C@H](O)[C@H]1O FZNCGRZWXLXZSZ-CIQUZCHMSA-N 0.000 description 1
- RRHHPXJSGSYSHW-ZURHXDBDSA-A [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].COC[C@H]1O[C@H](O[C@@H]2[C@@H](COC)O[C@@H](O[C@@H]3[C@@H](COC)O[C@H](O[C@@H]4[C@@H](COC)O[C@@H](O[C@H]5[C@@H](COS([O-])(=O)=O)O[C@H](O[C@H]6[C@H](OC)[C@@H](OC)[C@H](O[C@@H]7[C@@H](COS([O-])(=O)=O)O[C@H](O[C@H]8[C@H](OC)[C@H](OC)[C@H](O[C@@H]9[C@@H](COS([O-])(=O)=O)O[C@H](OC)[C@@H](OS([O-])(=O)=O)[C@H]9OC)O[C@H]8C([O-])=O)[C@H](OS([O-])(=O)=O)[C@H]7OS([O-])(=O)=O)O[C@H]6C([O-])=O)[C@H](OC)[C@H]5OC)[C@H](OC)[C@H]4OC)[C@H](OC)[C@H]3OC)[C@H](OC)[C@H]2OC)[C@H](OC)[C@@H](OC)[C@@H]1O[C@@H]1O[C@H](COC)[C@@H](O[C@@H]2O[C@H](COC)[C@@H](O[C@@H]3O[C@H](COC)[C@@H](O[C@H]4O[C@H](COS([O-])(=O)=O)[C@@H](O[C@@H]5O[C@H](COS([O-])(=O)=O)[C@@H](O[C@H]6O[C@H](COS([O-])(=O)=O)[C@@H](O[C@H]7O[C@H](COS([O-])(=O)=O)[C@@H](OS([O-])(=O)=O)[C@H](OS([O-])(=O)=O)[C@H]7OS([O-])(=O)=O)[C@H](OS([O-])(=O)=O)[C@H]6OS([O-])(=O)=O)[C@H](OS([O-])(=O)=O)[C@H]5OS([O-])(=O)=O)[C@H](OC)[C@H]4OC)[C@H](OC)[C@H]3OC)[C@H](OC)[C@H]2OC)[C@H](OC)[C@H]1OC Chemical compound [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].COC[C@H]1O[C@H](O[C@@H]2[C@@H](COC)O[C@@H](O[C@@H]3[C@@H](COC)O[C@H](O[C@@H]4[C@@H](COC)O[C@@H](O[C@H]5[C@@H](COS([O-])(=O)=O)O[C@H](O[C@H]6[C@H](OC)[C@@H](OC)[C@H](O[C@@H]7[C@@H](COS([O-])(=O)=O)O[C@H](O[C@H]8[C@H](OC)[C@H](OC)[C@H](O[C@@H]9[C@@H](COS([O-])(=O)=O)O[C@H](OC)[C@@H](OS([O-])(=O)=O)[C@H]9OC)O[C@H]8C([O-])=O)[C@H](OS([O-])(=O)=O)[C@H]7OS([O-])(=O)=O)O[C@H]6C([O-])=O)[C@H](OC)[C@H]5OC)[C@H](OC)[C@H]4OC)[C@H](OC)[C@H]3OC)[C@H](OC)[C@H]2OC)[C@H](OC)[C@@H](OC)[C@@H]1O[C@@H]1O[C@H](COC)[C@@H](O[C@@H]2O[C@H](COC)[C@@H](O[C@@H]3O[C@H](COC)[C@@H](O[C@H]4O[C@H](COS([O-])(=O)=O)[C@@H](O[C@@H]5O[C@H](COS([O-])(=O)=O)[C@@H](O[C@H]6O[C@H](COS([O-])(=O)=O)[C@@H](O[C@H]7O[C@H](COS([O-])(=O)=O)[C@@H](OS([O-])(=O)=O)[C@H](OS([O-])(=O)=O)[C@H]7OS([O-])(=O)=O)[C@H](OS([O-])(=O)=O)[C@H]6OS([O-])(=O)=O)[C@H](OS([O-])(=O)=O)[C@H]5OS([O-])(=O)=O)[C@H](OC)[C@H]4OC)[C@H](OC)[C@H]3OC)[C@H](OC)[C@H]2OC)[C@H](OC)[C@H]1OC RRHHPXJSGSYSHW-ZURHXDBDSA-A 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- BZKPWHYZMXOIDC-UHFFFAOYSA-N acetazolamide Chemical compound CC(=O)NC1=NN=C(S(N)(=O)=O)S1 BZKPWHYZMXOIDC-UHFFFAOYSA-N 0.000 description 1
- 229960000571 acetazolamide Drugs 0.000 description 1
- 229960001466 acetohexamide Drugs 0.000 description 1
- VGZSUPCWNCWDAN-UHFFFAOYSA-N acetohexamide Chemical compound C1=CC(C(=O)C)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 VGZSUPCWNCWDAN-UHFFFAOYSA-N 0.000 description 1
- 229960004308 acetylcysteine Drugs 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000009056 active transport Effects 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- MKOMESMZHZNBIZ-UHFFFAOYSA-M alagebrium Chemical compound [Cl-].CC1=C(C)SC=[N+]1CC(=O)C1=CC=CC=C1 MKOMESMZHZNBIZ-UHFFFAOYSA-M 0.000 description 1
- 229960004733 albiglutide Drugs 0.000 description 1
- OGWAVGNOAMXIIM-UHFFFAOYSA-N albiglutide Chemical compound O=C(O)C(NC(=O)CNC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)CNC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)CNC(=O)C(NC(=O)CNC(=O)C(N)CC=1(N=CNC=1))CCC(=O)O)C(O)C)CC2(=CC=CC=C2))C(O)C)CO)CC(=O)O)C(C)C)CO)CO)CC3(=CC=C(O)C=C3))CC(C)C)CCC(=O)O)CCC(=O)N)C)C)CCCCN)CCC(=O)O)CC4(=CC=CC=C4))C(CC)C)C)CC=6(C5(=C(C=CC=C5)NC=6)))CC(C)C)C(C)C)CCCCN)CCCNC(=N)N OGWAVGNOAMXIIM-UHFFFAOYSA-N 0.000 description 1
- 239000003288 aldose reductase inhibitor Substances 0.000 description 1
- 239000002170 aldosterone antagonist Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000002009 alkene group Chemical group 0.000 description 1
- 125000005210 alkyl ammonium group Chemical group 0.000 description 1
- 125000005119 alkyl cycloalkyl group Chemical group 0.000 description 1
- ZSBOMTDTBDDKMP-OAHLLOKOSA-N alogliptin Chemical compound C=1C=CC=C(C#N)C=1CN1C(=O)N(C)C(=O)C=C1N1CCC[C@@H](N)C1 ZSBOMTDTBDDKMP-OAHLLOKOSA-N 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- HFHDHCJBZVLPGP-RWMJIURBSA-N alpha-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO HFHDHCJBZVLPGP-RWMJIURBSA-N 0.000 description 1
- 229960003318 alteplase Drugs 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 description 1
- 229960000528 amlodipine Drugs 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 1
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 1
- ZRALSGWEFCBTJO-UHFFFAOYSA-N anhydrous guanidine Natural products NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 229940127003 anti-diabetic drug Drugs 0.000 description 1
- 230000001887 anti-feedant effect Effects 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 229940127218 antiplatelet drug Drugs 0.000 description 1
- 239000004019 antithrombin Substances 0.000 description 1
- 229960003886 apixaban Drugs 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 239000002830 appetite depressant Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- KXNPVXPOPUZYGB-XYVMCAHJSA-N argatroban Chemical compound OC(=O)[C@H]1C[C@H](C)CCN1C(=O)[C@H](CCCN=C(N)N)NS(=O)(=O)C1=CC=CC2=C1NC[C@H](C)C2 KXNPVXPOPUZYGB-XYVMCAHJSA-N 0.000 description 1
- 229960003856 argatroban Drugs 0.000 description 1
- 208000021328 arterial occlusion Diseases 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 210000000467 autonomic pathway Anatomy 0.000 description 1
- 229950010046 avasimibe Drugs 0.000 description 1
- KGSXMPPBFPAXLY-UHFFFAOYSA-N azilsartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NOC(=O)N1 KGSXMPPBFPAXLY-UHFFFAOYSA-N 0.000 description 1
- 125000005604 azodicarboxylate group Chemical group 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 1
- QQFYLZXBFWWJHR-UHFFFAOYSA-M benzyl(triethyl)phosphanium;bromide Chemical compound [Br-].CC[P+](CC)(CC)CC1=CC=CC=C1 QQFYLZXBFWWJHR-UHFFFAOYSA-M 0.000 description 1
- 229960002890 beraprost Drugs 0.000 description 1
- YTCZZXIRLARSET-VJRSQJMHSA-M beraprost sodium Chemical compound [Na+].O([C@H]1C[C@@H](O)[C@@H]([C@@H]21)/C=C/[C@@H](O)C(C)CC#CC)C1=C2C=CC=C1CCCC([O-])=O YTCZZXIRLARSET-VJRSQJMHSA-M 0.000 description 1
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 description 1
- 229960004853 betadex Drugs 0.000 description 1
- 229960000516 bezafibrate Drugs 0.000 description 1
- IIBYAHWJQTYFKB-UHFFFAOYSA-N bezafibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1CCNC(=O)C1=CC=C(Cl)C=C1 IIBYAHWJQTYFKB-UHFFFAOYSA-N 0.000 description 1
- 210000003445 biliary tract Anatomy 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 108010055460 bivalirudin Proteins 0.000 description 1
- OIRCOABEOLEUMC-GEJPAHFPSA-N bivalirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)CNC(=O)CNC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 OIRCOABEOLEUMC-GEJPAHFPSA-N 0.000 description 1
- 229960001500 bivalirudin Drugs 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 229960004111 buformin Drugs 0.000 description 1
- XSEUMFJMFFMCIU-UHFFFAOYSA-N buformin Chemical compound CCCC\N=C(/N)N=C(N)N XSEUMFJMFFMCIU-UHFFFAOYSA-N 0.000 description 1
- 239000004067 bulking agent Substances 0.000 description 1
- IAQRGUVFOMOMEM-UHFFFAOYSA-N butene Natural products CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 description 1
- 235000014121 butter Nutrition 0.000 description 1
- AVVIDTZRJBSXML-UHFFFAOYSA-L calcium;2-carboxyphenolate;dihydrate Chemical compound O.O.[Ca+2].OC1=CC=CC=C1C([O-])=O.OC1=CC=CC=C1C([O-])=O AVVIDTZRJBSXML-UHFFFAOYSA-L 0.000 description 1
- ZQNPDAVSHFGLIQ-UHFFFAOYSA-N calcium;hydrate Chemical compound O.[Ca] ZQNPDAVSHFGLIQ-UHFFFAOYSA-N 0.000 description 1
- 229960004349 candesartan cilexetil Drugs 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 description 1
- 239000003489 carbonate dehydratase inhibitor Substances 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 229940084030 carboxymethylcellulose calcium Drugs 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 208000026106 cerebrovascular disease Diseases 0.000 description 1
- MVCQKIKWYUURMU-UHFFFAOYSA-N cetilistat Chemical compound C1=C(C)C=C2C(=O)OC(OCCCCCCCCCCCCCCCC)=NC2=C1 MVCQKIKWYUURMU-UHFFFAOYSA-N 0.000 description 1
- 229950002397 cetilistat Drugs 0.000 description 1
- JUFFVKRROAPVBI-PVOYSMBESA-N chembl1210015 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N[C@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@]3(O[C@@H](C[C@H](O)[C@H](O)CO)[C@H](NC(C)=O)[C@@H](O)C3)C(O)=O)O2)O)[C@@H](CO)O1)NC(C)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 JUFFVKRROAPVBI-PVOYSMBESA-N 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- VXIVSQZSERGHQP-UHFFFAOYSA-N chloroacetamide Chemical compound NC(=O)CCl VXIVSQZSERGHQP-UHFFFAOYSA-N 0.000 description 1
- 229960001761 chlorpropamide Drugs 0.000 description 1
- 229960001523 chlortalidone Drugs 0.000 description 1
- 230000001906 cholesterol absorption Effects 0.000 description 1
- 239000003354 cholesterol ester transfer protein inhibitor Substances 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 230000001886 ciliary effect Effects 0.000 description 1
- 229960004588 cilostazol Drugs 0.000 description 1
- RRGUKTPIGVIEKM-UHFFFAOYSA-N cilostazol Chemical compound C=1C=C2NC(=O)CCC2=CC=1OCCCCC1=NN=NN1C1CCCCC1 RRGUKTPIGVIEKM-UHFFFAOYSA-N 0.000 description 1
- 229950003072 clinofibrate Drugs 0.000 description 1
- 229960001214 clofibrate Drugs 0.000 description 1
- KNHUKKLJHYUCFP-UHFFFAOYSA-N clofibrate Chemical compound CCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 KNHUKKLJHYUCFP-UHFFFAOYSA-N 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 210000002858 crystal cell Anatomy 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 229960003206 cyclopenthiazide Drugs 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 229960003850 dabigatran Drugs 0.000 description 1
- YBSJFWOBGCMAKL-UHFFFAOYSA-N dabigatran Chemical compound N=1C2=CC(C(=O)N(CCC(O)=O)C=3N=CC=CC=3)=CC=C2N(C)C=1CNC1=CC=C(C(N)=N)C=C1 YBSJFWOBGCMAKL-UHFFFAOYSA-N 0.000 description 1
- 229940018872 dalteparin sodium Drugs 0.000 description 1
- 229960003834 dapagliflozin Drugs 0.000 description 1
- 229960005227 delapril Drugs 0.000 description 1
- WOUOLAUOZXOLJQ-MBSDFSHPSA-N delapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N(CC(O)=O)C1CC2=CC=CC=C2C1)CC1=CC=CC=C1 WOUOLAUOZXOLJQ-MBSDFSHPSA-N 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 108010073652 desirudin Proteins 0.000 description 1
- XYWBJDRHGNULKG-OUMQNGNKSA-N desirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H]1NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H]2CSSC[C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@H](C(NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N2)=O)CSSC1)C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)[C@@H](C)O)CSSC1)C(C)C)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 XYWBJDRHGNULKG-OUMQNGNKSA-N 0.000 description 1
- 229960000296 desirudin Drugs 0.000 description 1
- 229960004597 dexfenfluramine Drugs 0.000 description 1
- 208000033679 diabetic kidney disease Diseases 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- 229960004890 diethylpropion Drugs 0.000 description 1
- XXEPPPIWZFICOJ-UHFFFAOYSA-N diethylpropion Chemical compound CCN(CC)C(C)C(=O)C1=CC=CC=C1 XXEPPPIWZFICOJ-UHFFFAOYSA-N 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 239000003603 dipeptidyl peptidase IV inhibitor Substances 0.000 description 1
- BTLCTWYLBFXODL-UHFFFAOYSA-N diphenyl(2H-pyridin-1-yl)phosphane Chemical compound C1(=CC=CC=C1)P(N1CC=CC=C1)C1=CC=CC=C1 BTLCTWYLBFXODL-UHFFFAOYSA-N 0.000 description 1
- SVABQOITNJTVNJ-UHFFFAOYSA-N diphenyl-2-pyridylphosphine Chemical compound C1=CC=CC=C1P(C=1N=CC=CC=1)C1=CC=CC=C1 SVABQOITNJTVNJ-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 229950005925 eflucimibe Drugs 0.000 description 1
- 229950003102 efonidipine Drugs 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229960004563 eprosartan Drugs 0.000 description 1
- OROAFUQRIXKEMV-LDADJPATSA-N eprosartan Chemical compound C=1C=C(C(O)=O)C=CC=1CN1C(CCCC)=NC=C1\C=C(C(O)=O)/CC1=CC=CS1 OROAFUQRIXKEMV-LDADJPATSA-N 0.000 description 1
- AVOLMBLBETYQHX-UHFFFAOYSA-N etacrynic acid Chemical compound CCC(=C)C(=O)C1=CC=C(OCC(O)=O)C(Cl)=C1Cl AVOLMBLBETYQHX-UHFFFAOYSA-N 0.000 description 1
- 229960003199 etacrynic acid Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 229950007164 ethiazide Drugs 0.000 description 1
- SSQPWTVBQMWLSZ-AAQCHOMXSA-N ethyl (5Z,8Z,11Z,14Z,17Z)-icosapentaenoate Chemical compound CCOC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CC SSQPWTVBQMWLSZ-AAQCHOMXSA-N 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000009207 exercise therapy Methods 0.000 description 1
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 1
- 229940125753 fibrate Drugs 0.000 description 1
- 239000003527 fibrinolytic agent Substances 0.000 description 1
- WAAPEIZFCHNLKK-PELKAZGASA-N fidarestat Chemical compound C([C@@H](OC1=CC=C(F)C=C11)C(=O)N)[C@@]21NC(=O)NC2=O WAAPEIZFCHNLKK-PELKAZGASA-N 0.000 description 1
- 229950007256 fidarestat Drugs 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 229960001318 fondaparinux Drugs 0.000 description 1
- KANJSNBRCNMZMV-ABRZTLGGSA-N fondaparinux Chemical compound O[C@@H]1[C@@H](NS(O)(=O)=O)[C@@H](OC)O[C@H](COS(O)(=O)=O)[C@H]1O[C@H]1[C@H](OS(O)(=O)=O)[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](OS(O)(=O)=O)[C@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O[C@@H]4[C@@H]([C@@H](O)[C@H](O)[C@@H](COS(O)(=O)=O)O4)NS(O)(=O)=O)[C@H](O3)C(O)=O)O)[C@@H](COS(O)(=O)=O)O2)NS(O)(=O)=O)[C@H](C(O)=O)O1 KANJSNBRCNMZMV-ABRZTLGGSA-N 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- FATAVLOOLIRUNA-UHFFFAOYSA-N formylmethyl Chemical group [CH2]C=O FATAVLOOLIRUNA-UHFFFAOYSA-N 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 229960003883 furosemide Drugs 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- FODTZLFLDFKIQH-FSVGXZBPSA-N gamma-Oryzanol (TN) Chemical compound C1=C(O)C(OC)=CC(\C=C\C(=O)O[C@@H]2C([C@@H]3CC[C@H]4[C@]5(C)CC[C@@H]([C@@]5(C)CC[C@@]54C[C@@]53CC2)[C@H](C)CCC=C(C)C)(C)C)=C1 FODTZLFLDFKIQH-FSVGXZBPSA-N 0.000 description 1
- GDSRMADSINPKSL-HSEONFRVSA-N gamma-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO GDSRMADSINPKSL-HSEONFRVSA-N 0.000 description 1
- 229940080345 gamma-cyclodextrin Drugs 0.000 description 1
- GNKDKYIHGQKHHM-RJKLHVOGSA-N ghrelin Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)CN)COC(=O)CCCCCCC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C1=CC=CC=C1 GNKDKYIHGQKHHM-RJKLHVOGSA-N 0.000 description 1
- 229960004346 glimepiride Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical compound O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 1
- 229960004666 glucagon Drugs 0.000 description 1
- 239000003877 glucagon like peptide 1 receptor agonist Substances 0.000 description 1
- 230000004153 glucose metabolism Effects 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- MNQZXJOMYWMBOU-UHFFFAOYSA-N glyceraldehyde Chemical compound OCC(O)C=O MNQZXJOMYWMBOU-UHFFFAOYSA-N 0.000 description 1
- 229930182470 glycoside Natural products 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- ZFGMDIBRIDKWMY-PASTXAENSA-N heparin Chemical compound CC(O)=N[C@@H]1[C@@H](O)[C@H](O)[C@@H](COS(O)(=O)=O)O[C@@H]1O[C@@H]1[C@@H](C(O)=O)O[C@@H](O[C@H]2[C@@H]([C@@H](OS(O)(=O)=O)[C@@H](O[C@@H]3[C@@H](OC(O)[C@H](OS(O)(=O)=O)[C@H]3O)C(O)=O)O[C@@H]2O)CS(O)(=O)=O)[C@H](O)[C@H]1O ZFGMDIBRIDKWMY-PASTXAENSA-N 0.000 description 1
- 229940095529 heparin calcium Drugs 0.000 description 1
- 229960001008 heparin sodium Drugs 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- DMDGGSIALPNSEE-UHFFFAOYSA-N hydroflumethiazide Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O DMDGGSIALPNSEE-UHFFFAOYSA-N 0.000 description 1
- 229960003313 hydroflumethiazide Drugs 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- GRRNUXAQVGOGFE-NZSRVPFOSA-N hygromycin B Chemical compound O[C@@H]1[C@@H](NC)C[C@@H](N)[C@H](O)[C@H]1O[C@H]1[C@H]2O[C@@]3([C@@H]([C@@H](O)[C@@H](O)[C@@H](C(N)CO)O3)O)O[C@H]2[C@@H](O)[C@@H](CO)O1 GRRNUXAQVGOGFE-NZSRVPFOSA-N 0.000 description 1
- 229940097277 hygromycin b Drugs 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 230000003451 hyperinsulinaemic effect Effects 0.000 description 1
- 201000008980 hyperinsulinism Diseases 0.000 description 1
- 208000006575 hypertriglyceridemia Diseases 0.000 description 1
- 229960004569 indapamide Drugs 0.000 description 1
- NDDAHWYSQHTHNT-UHFFFAOYSA-N indapamide Chemical compound CC1CC2=CC=CC=C2N1NC(=O)C1=CC=C(Cl)C(S(N)(=O)=O)=C1 NDDAHWYSQHTHNT-UHFFFAOYSA-N 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- PBGKTOXHQIOBKM-FHFVDXKLSA-N insulin (human) Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3C=CC(O)=CC=3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3NC=NC=3)NC(=O)[C@H](CO)NC(=O)CNC1=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)=O)CSSC[C@@H](C(N2)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)[C@@H](C)CC)[C@@H](C)O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C1=CN=CN1 PBGKTOXHQIOBKM-FHFVDXKLSA-N 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000002510 isobutoxy group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])O* 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 229960002479 isosorbide Drugs 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- OTQCKZUSUGYWBD-BRHMIFOHSA-N lepirudin Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)C(C)C)[C@@H](C)O)[C@@H](C)O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC(C)C)[C@@H](C)O)C1=CC=C(O)C=C1 OTQCKZUSUGYWBD-BRHMIFOHSA-N 0.000 description 1
- 229960004408 lepirudin Drugs 0.000 description 1
- 229940039781 leptin Drugs 0.000 description 1
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- 229960002701 liraglutide Drugs 0.000 description 1
- 229940083747 low-ceiling diuretics xanthine derivative Drugs 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 150000002689 maleic acids Chemical class 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229960000299 mazindol Drugs 0.000 description 1
- 235000012054 meals Nutrition 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 1
- FWMHZWMPUWAUPL-NDEPHWFRSA-N methyl (4s)-3-[3-[4-(3-acetamidophenyl)piperidin-1-yl]propylcarbamoyl]-4-(3,4-difluorophenyl)-6-(methoxymethyl)-2-oxo-1,4-dihydropyrimidine-5-carboxylate Chemical compound N1([C@H](C(=C(NC1=O)COC)C(=O)OC)C=1C=C(F)C(F)=CC=1)C(=O)NCCCN(CC1)CCC1C1=CC=CC(NC(C)=O)=C1 FWMHZWMPUWAUPL-NDEPHWFRSA-N 0.000 description 1
- 229960003404 mexiletine Drugs 0.000 description 1
- 206010062198 microangiopathy Diseases 0.000 description 1
- 229960001110 miglitol Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 229960003365 mitiglinide Drugs 0.000 description 1
- WPGGHFDDFPHPOB-BBWFWOEESA-N mitiglinide Chemical compound C([C@@H](CC(=O)N1C[C@@H]2CCCC[C@@H]2C1)C(=O)O)C1=CC=CC=C1 WPGGHFDDFPHPOB-BBWFWOEESA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 108010075698 monteplase Proteins 0.000 description 1
- 229950005805 monteplase Drugs 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- VYNKVNDKAOGAAQ-RUZDIDTESA-N n-[(1r)-2-[4-(1-methylpiperidin-4-yl)piperazin-1-yl]-2-oxo-1-phenylethyl]-1h-indole-6-carboxamide Chemical compound C1CN(C)CCC1N1CCN(C(=O)[C@H](NC(=O)C=2C=C3NC=CC3=CC=2)C=2C=CC=CC=2)CC1 VYNKVNDKAOGAAQ-RUZDIDTESA-N 0.000 description 1
- 125000006606 n-butoxy group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- BDQCDIWFPIDPQU-NDEPHWFRSA-N n-methyl-5-[4-[1-[(1r)-3-oxospiro[2-benzofuran-1,3'-pyrrolidine]-1'-carbonyl]cyclopropyl]phenyl]pyridine-2-carboxamide Chemical compound C1=NC(C(=O)NC)=CC=C1C1=CC=C(C2(CC2)C(=O)N2C[C@@]3(CC2)C2=CC=CC=C2C(=O)O3)C=C1 BDQCDIWFPIDPQU-NDEPHWFRSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229950003494 naveglitazar Drugs 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 230000000508 neurotrophic effect Effects 0.000 description 1
- 239000003900 neurotrophic factor Substances 0.000 description 1
- 230000032064 neurotrophin production Effects 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 229960000827 niceritrol Drugs 0.000 description 1
- 229950001071 nicomol Drugs 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- MVPQUSQUURLQKF-MCPDASDXSA-E nonasodium;(2s,3s,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3s,4s,5r,6r)-2-carboxylato-4,5-dimethoxy-6-[(2r,3r,4s,5r,6s)-6-methoxy-4,5-disulfonatooxy-2-(sulfonatooxymethyl)oxan-3-yl]oxyoxan-3-yl]oxy-4,5-disulfonatooxy-2-(sulfonatooxymethyl)oxan-3-yl]oxy-4,5-di Chemical compound [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[O-]S(=O)(=O)O[C@@H]1[C@@H](OS([O-])(=O)=O)[C@@H](OC)O[C@H](COS([O-])(=O)=O)[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@@H]2[C@@H]([C@@H](OS([O-])(=O)=O)[C@H](O[C@H]3[C@@H]([C@@H](OC)[C@H](O[C@@H]4[C@@H]([C@@H](OC)[C@H](OC)[C@@H](COS([O-])(=O)=O)O4)OC)[C@H](O3)C([O-])=O)OC)[C@@H](COS([O-])(=O)=O)O2)OS([O-])(=O)=O)[C@H](C([O-])=O)O1 MVPQUSQUURLQKF-MCPDASDXSA-E 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 229950005036 odiparcil Drugs 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 229940125395 oral insulin Drugs 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- PFGVNLZDWRZPJW-OPAMFIHVSA-N otamixaban Chemical compound C([C@@H](C(=O)OC)[C@@H](C)NC(=O)C=1C=CC(=CC=1)C=1C=C[N+]([O-])=CC=1)C1=CC=CC(C(N)=N)=C1 PFGVNLZDWRZPJW-OPAMFIHVSA-N 0.000 description 1
- 229950009478 otamixaban Drugs 0.000 description 1
- JRTYPQGPARWINR-UHFFFAOYSA-N palladium platinum Chemical compound [Pd].[Pt] JRTYPQGPARWINR-UHFFFAOYSA-N 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- 108010085108 pamiteplase Proteins 0.000 description 1
- 229950003603 pamiteplase Drugs 0.000 description 1
- MFPRYDMAZLLTGM-IJDGSQHYSA-N pegmusirudin Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC1=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@H](C(NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC1=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCNC(=O)OCCOCC)C(=O)N2)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)=O)CCCCNC(=O)OCCOCC)[C@@H](C)CC)[C@@H](C)O)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)C1=CC=C(O)C=C1 MFPRYDMAZLLTGM-IJDGSQHYSA-N 0.000 description 1
- 229950001482 pegmusirudin Drugs 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 229960003243 phenformin Drugs 0.000 description 1
- ICFJFFQQTFMIBG-UHFFFAOYSA-N phenformin Chemical compound NC(=N)NC(=N)NCCC1=CC=CC=C1 ICFJFFQQTFMIBG-UHFFFAOYSA-N 0.000 description 1
- 229960003562 phentermine Drugs 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 125000000109 phenylethoxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])O* 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000003003 phosphines Chemical class 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 235000002378 plant sterols Nutrition 0.000 description 1
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229960005483 polythiazide Drugs 0.000 description 1
- 229920000046 polythiazide Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 229960003611 pramlintide Drugs 0.000 description 1
- 108010029667 pramlintide Proteins 0.000 description 1
- NRKVKVQDUCJPIZ-MKAGXXMWSA-N pramlintide acetate Chemical compound C([C@@H](C(=O)NCC(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CS)NC(=O)[C@@H](N)CCCCN)[C@@H](C)O)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 NRKVKVQDUCJPIZ-MKAGXXMWSA-N 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- XTRUQJBVQBUKSQ-UHFFFAOYSA-N propan-2-yl 4-[1-(2-fluoro-4-methylsulfonylphenyl)pyrazolo[3,4-d]pyrimidin-4-yl]oxypiperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)C)CCC1OC1=NC=NC2=C1C=NN2C1=CC=C(S(C)(=O)=O)C=C1F XTRUQJBVQBUKSQ-UHFFFAOYSA-N 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 229940048914 protamine Drugs 0.000 description 1
- LHZWKWCEAXQUMX-UHFFFAOYSA-N psn-632,408 Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1OCC1=NC(C=2C=CN=CC=2)=NO1 LHZWKWCEAXQUMX-UHFFFAOYSA-N 0.000 description 1
- JEXVQSWXXUJEMA-UHFFFAOYSA-N pyrazol-3-one Chemical compound O=C1C=CN=N1 JEXVQSWXXUJEMA-UHFFFAOYSA-N 0.000 description 1
- HDOUGSFASVGDCS-UHFFFAOYSA-N pyridin-3-ylmethanamine Chemical compound NCC1=CC=CN=C1 HDOUGSFASVGDCS-UHFFFAOYSA-N 0.000 description 1
- 235000008151 pyridoxamine Nutrition 0.000 description 1
- 239000011699 pyridoxamine Substances 0.000 description 1
- HNWCOANXZNKMLR-UHFFFAOYSA-N pyridoxamine dihydrochloride Chemical compound Cl.Cl.CC1=NC=C(CO)C(CN)=C1O HNWCOANXZNKMLR-UHFFFAOYSA-N 0.000 description 1
- 229950010535 razaxaban Drugs 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- UAOCLDQAQNNEAX-ABMICEGHSA-N remogliflozin etabonate Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](COC(=O)OCC)O[C@H]1OC1=NN(C(C)C)C(C)=C1CC1=CC=C(OC(C)C)C=C1 UAOCLDQAQNNEAX-ABMICEGHSA-N 0.000 description 1
- 230000030558 renal glucose absorption Effects 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- JZCPYUJPEARBJL-UHFFFAOYSA-N rimonabant Chemical compound CC=1C(C(=O)NN2CCCCC2)=NN(C=2C(=CC(Cl)=CC=2)Cl)C=1C1=CC=C(Cl)C=C1 JZCPYUJPEARBJL-UHFFFAOYSA-N 0.000 description 1
- 229960001148 rivaroxaban Drugs 0.000 description 1
- KGFYHTZWPPHNLQ-AWEZNQCLSA-N rivaroxaban Chemical compound S1C(Cl)=CC=C1C(=O)NC[C@@H]1OC(=O)N(C=2C=CC(=CC=2)N2C(COCC2)=O)C1 KGFYHTZWPPHNLQ-AWEZNQCLSA-N 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 108010033693 saxagliptin Proteins 0.000 description 1
- 229960004937 saxagliptin Drugs 0.000 description 1
- QGJUIPDUBHWZPV-SGTAVMJGSA-N saxagliptin Chemical compound C1C(C2)CC(C3)CC2(O)CC13[C@H](N)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QGJUIPDUBHWZPV-SGTAVMJGSA-N 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000001953 sensory effect Effects 0.000 description 1
- 229940126842 sergliflozin Drugs 0.000 description 1
- HFLCZNNDZKKXCS-OUUBHVDSSA-N sergliflozin Chemical compound C1=CC(OC)=CC=C1CC1=CC=CC=C1O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HFLCZNNDZKKXCS-OUUBHVDSSA-N 0.000 description 1
- 229960004425 sibutramine Drugs 0.000 description 1
- UNAANXDKBXWMLN-UHFFFAOYSA-N sibutramine Chemical compound C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UNAANXDKBXWMLN-UHFFFAOYSA-N 0.000 description 1
- 238000007873 sieving Methods 0.000 description 1
- 229960004058 simfibrate Drugs 0.000 description 1
- 229950005481 sipoglitazar Drugs 0.000 description 1
- 229960004034 sitagliptin Drugs 0.000 description 1
- MFFMDFFZMYYVKS-SECBINFHSA-N sitagliptin Chemical compound C([C@H](CC(=O)N1CC=2N(C(=NN=2)C(F)(F)F)CC1)N)C1=CC(F)=C(F)C=C1F MFFMDFFZMYYVKS-SECBINFHSA-N 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 210000003625 skull Anatomy 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- CMZUMMUJMWNLFH-UHFFFAOYSA-N sodium metavanadate Chemical compound [Na+].[O-][V](=O)=O CMZUMMUJMWNLFH-UHFFFAOYSA-N 0.000 description 1
- 229960004025 sodium salicylate Drugs 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 229940033331 soy sterol Drugs 0.000 description 1
- 239000004059 squalene synthase inhibitor Substances 0.000 description 1
- FWMUJAIKEJWSSY-UHFFFAOYSA-N sulfur dichloride Chemical compound ClSCl FWMUJAIKEJWSSY-UHFFFAOYSA-N 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000007916 tablet composition Substances 0.000 description 1
- 108010009573 talabostat Proteins 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 235000012976 tarts Nutrition 0.000 description 1
- 229960000651 tasosartan Drugs 0.000 description 1
- ADXGNEYLLLSOAR-UHFFFAOYSA-N tasosartan Chemical compound C12=NC(C)=NC(C)=C2CCC(=O)N1CC(C=C1)=CC=C1C1=CC=CC=C1C=1N=NNN=1 ADXGNEYLLLSOAR-UHFFFAOYSA-N 0.000 description 1
- WRGVLTAWMNZWGT-VQSPYGJZSA-N taspoglutide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NC(C)(C)C(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)C(C)(C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 WRGVLTAWMNZWGT-VQSPYGJZSA-N 0.000 description 1
- 229950007151 taspoglutide Drugs 0.000 description 1
- 229960003080 taurine Drugs 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- WGRQANOPCQRCME-PMACEKPBSA-N teneligliptin Chemical compound O=C([C@H]1NC[C@H](C1)N1CCN(CC1)C1=CC(=NN1C=1C=CC=CC=1)C)N1CCSC1 WGRQANOPCQRCME-PMACEKPBSA-N 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005505 thiomorpholino group Chemical group 0.000 description 1
- 229960002277 tolazamide Drugs 0.000 description 1
- OUDSBRTVNLOZBN-UHFFFAOYSA-N tolazamide Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1CCCCCC1 OUDSBRTVNLOZBN-UHFFFAOYSA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 1
- CMSGWTNRGKRWGS-NQIIRXRSSA-N torcetrapib Chemical compound COC(=O)N([C@H]1C[C@@H](CC)N(C2=CC=C(C=C21)C(F)(F)F)C(=O)OCC)CC1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 CMSGWTNRGKRWGS-NQIIRXRSSA-N 0.000 description 1
- 229950004514 torcetrapib Drugs 0.000 description 1
- 229960001288 triamterene Drugs 0.000 description 1
- LMJSLTNSBFUCMU-UHFFFAOYSA-N trichlormethiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NC(C(Cl)Cl)NS2(=O)=O LMJSLTNSBFUCMU-UHFFFAOYSA-N 0.000 description 1
- 229960004813 trichlormethiazide Drugs 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 1
- 208000019206 urinary tract infection Diseases 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 1
- 229940124549 vasodilator Drugs 0.000 description 1
- 239000003071 vasodilator agent Substances 0.000 description 1
- SYOKIDBDQMKNDQ-XWTIBIIYSA-N vildagliptin Chemical compound C1C(O)(C2)CC(C3)CC1CC32NCC(=O)N1CCC[C@H]1C#N SYOKIDBDQMKNDQ-XWTIBIIYSA-N 0.000 description 1
- 229960001729 voglibose Drugs 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- SXONDGSPUVNZLO-UHFFFAOYSA-N zenarestat Chemical compound O=C1N(CC(=O)O)C2=CC(Cl)=CC=C2C(=O)N1CC1=CC=C(Br)C=C1F SXONDGSPUVNZLO-UHFFFAOYSA-N 0.000 description 1
- 229950006343 zenarestat Drugs 0.000 description 1
- 229910000166 zirconium phosphate Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/02—Heterocyclic radicals containing only nitrogen as ring hetero atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to a pyrazolyl 5-thiodarcoside compound having an inhibitory activity on sodium-dependent glucose cotransporter 1 (SGLT1) involved in glucose reabsorption in the kidney.
- SGLT1 sodium-dependent glucose cotransporter 1
- the fasting blood glucose level is 126 mgZdL or more.
- IGT impaired glucose tolerance
- Non-Patent Document 2 administration of the a-darcosidase inhibitor acarbose, which inhibits sugar hydrolase and delays the absorption of sugar in the small intestine, suppresses the transition from IGT to type 2 diabetes. Furthermore, it has been reported that the onset of hypertension is also significantly suppressed (see Non-Patent Document 2).
- SG LT1 Sodium-dependent glucose cotransporter 1
- IGT Sodium-dependent glucose cotransporter 1
- Non-Patent Documents sodium-dependent dalcose cotransporter 2 (SGLT2) is frequently expressed in the kidney, and glucose that has been glomerularly filtered is reabsorbed via SGLT2 (Non-Patent Documents). 3). SGLT2 inhibitors promote glucose excretion in the urine and reduce blood sugar. As a result, it has come to be considered as a target molecule for new antidiabetic drugs (see Non-Patent Document 4). against this background, SGLT2 inhibitors have been studied and heteroaryl 5-thiodarcoside derivatives have been provided (see Patent Document 7).
- Patent Document 1 International Publication No. WO2002Z098893 Pamphlet
- Patent Document 2 International Publication No. WO2004Z014932 Pamphlet
- Patent Document 3 International Publication No. WO2004Z018491 Pamphlet
- Patent Document 4 International Publication No. WO2004Z019958 Pamphlet
- Patent Document 5 International Publication No. WO2005Z121161 Pamphlet
- Patent Document 6 International Publication No. WO2004Z050122 Pamphlet
- Patent Document 7 International Publication No. WO2004Z089967 Pamphlet
- Non-Patent Document 1 Pan XR, et al. Diabets Care, VIII, 534, 1997
- Non-Patent Document 2 J.-Shi Chiasson, et al. Lancent, 359, 2072, 2002
- Non-Patent Document 3 E. M. Wright, Am. J. Physiol. Renal. Physiol., 280, F10, 2001
- Non-Patent Document 4 G. Toggenburger, et al. Biochem. Biophys. Acta., 688, 557, 1 982
- the present invention provides a novel pyrazolyl 5-thio-13D-darcoviranoside compound that controls IGT by selectively inhibiting SGLT1 activity and suppressing glucose absorption from the gastrointestinal tract. Is an issue.
- the pyrazolyl 5 thiodarcoside derivative of the present invention (hereinafter referred to as "the compound of the present invention”) )) Is described.
- the embodiment (1) of the present invention is a pyrazolyl 5-thiodarcoside compound represented by the following formula (I), a pharmaceutically acceptable salt thereof, or a hydrate thereof.
- Z is a C alkyl group
- Y is a single bond, a C alkylene group, a C alkylene group or —O— (CH) n— (n is 1
- Q is a hydroxyl group, -OC alkyl file, -N (R A ) R B or CONHRC,
- R A and R B may be the same or different and may be substituted with a hydrogen atom, —CONHR D (R D is a hydrogen atom or a substituent selected from the group consisting of a hydroxyl group and a pyridyl group!
- R F and R are the same or different and may be a hydrogen atom or a C alkyl group
- R F and R a together with the nitrogen atom to which they are attached connexion, a further ring-constituting atom, an oxygen atom, a sulfur atom and to the to be good 5-6 membered comprise hetero atoms selected from nitrogen atom Telocycloalkyl group (the heterocycloalkyl group may be substituted with a hydroxyl group)
- V, C may be substituted with alkyl groups, may form)! / ,.
- the dotted line of the pyrazole ring indicates that a tautomer exists depending on the position where the hydrogen atom is bonded to the ring nitrogen atom. For convenience, in the specific compound shown below, Only one of the isomers is shown.
- Y is a C alkylene group, a C alkylene group or —O—.
- Another embodiment of the present invention is the embodiment (1) or (2), wherein Q is -N (R A ) R B (R A and R B are as defined in embodiment (1)). Or a pharmaceutically acceptable salt thereof or a hydrate thereof.
- R A is —CONHR D (wherein R D is a C alkyl group substituted with a substituent selected from the group consisting of a hydroxyl group and a pyridyl group), R B is water
- R A is —C ( ⁇ NH) NHR E (R E is a hydrogen atom, or —CO
- C. is an alkylphenyl
- R B is a hydrogen atom
- R A and R B are the same or different, and a hydrogen atom or C
- Luca may also be substituted with a selected substituent! )
- Another embodiment of the present invention is a pyrazolyl 5-thiodarcosidoxy compound according to embodiment (1) or (2), wherein Q is -CONHR e (R e is as defined in embodiment (1)).
- Q is -CONHR e (R e is as defined in embodiment (1)).
- R E is selected from the group consisting of a hydroxyl group and —CONH key 1
- the piperazino may be substituted with a C alkyl group optionally substituted with a hydroxyl group
- R G is each C alkyl bonded to —NH—
- the carbon atom of 1-6 group is tertiary), and the pyrazolyl 5-thiodarcoside compound according to embodiment (7) or a pharmaceutically acceptable salt thereof or a hydrate thereof.
- Y is a C alkylene group or a C alkene group.
- Y is O— (CH 2) n- (n is an integer from 1 to 4);
- Q is a hydroxyl group, an OC alkylphenyl, or —N (R A ) R B ;
- R A and R B are the same or different and are each a hydrogen atom, C alkyl substituted with -CONH.
- R D is a C alkyl group substituted with a hydroxyl group
- the pyrazolyl 5-thiodarcoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof according to any one of embodiments (1) to (10) is effective.
- SGLT1 activity inhibitor contained as a component.
- the pyrazolyl 5-thiodarcoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof according to any one of embodiments (1) to (10) is effective. It is a preventive or therapeutic agent for diabetes included as a component.
- the compound of the present invention selectively inhibits SGLT1 activity, borderline type (diabetic It is possible to treat patients in the group), thereby preventing the transition to diabetes.
- C alkyl group means a linear or branched alkyl group having 1 to 6 carbon atoms.
- a methyl group, an ethyl group, an n propyl group, an isopropyl group, an n-butyl group, an isobutyl group, a tert butyl group, a sec butyl group, an n pentyl group, and an n-hexyl group can be mentioned.
- C alkoxy group means a straight-chain or branched alkoxy having 16 carbon atoms.
- Means a Si group, and a C alkoxy group is preferred.
- Examples of the C alkoxy group include meth
- halogen atom is a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- One OC alkyl file has a form in which a C alkoxy group and a file are combined.
- the "C alkylene group” is a linear or branched divalent saturated group having 1 to 6 carbon atoms.
- hydrocarbon It is a hydrocarbon.
- methylene, ethylene, trimethylene, tetramethylene, 2-methylpropylene, 2,2-dimethylpropylene and the like can be mentioned.
- the "C alk-lene group” is a straight chain or branched chain having 2 to 6 carbon atoms including a double bond.
- ethylene, probelene, 2-butylene and the like can be mentioned. Of these, probelene is preferable.
- C-a which may be substituted with a substituent selected from the group consisting of a hydroxyl group and a pyridyl group
- 1-6 alkyl group means that the hydrogen atom on the c alkyl group is selected from a hydroxyl group and a pyridyl group.
- a C alkyl group optionally substituted by one or more groups (preferably 1 to 3)
- methyl group, ethyl group, isopropyl group, pyridine 3-ylmethyl group, hydroxymethyl group, hydroxyethyl group, 2-hydroxy-1,1-dimethylethyl group, 1,3-dihydroxy-2 hydroxymethylpropane-2-yl group Can be mentioned.
- substitution selected from the group consisting of hydroxyl group, -CONH and OC alkylfur C alkyl group optionally substituted with a group means that a hydrogen atom on a C alkyl group is a hydroxyl group.
- One or more selected from the group consisting of the groups CONH and OC alkylphenyl preferably
- C represents an alkyl group which may be preferably substituted by 1 to 3 groups.
- Mechi represents an alkyl group which may be preferably substituted by 1 to 3 groups.
- ethyl group isopropyl group, benzyloxycetyl group, hydroxymethyl group, hydroxyxetyl group, 2 hydroxy 1,1-dimethylethyl group, 1,3 hydroxy-2-methylpropane 2-yl group, strong rubamoylmethyl group, Examples thereof include a strong ruberamoylmethyl group, a 2-carbamoylethyl group, and a 2-force rubamoyl-2-methylethyl group.
- 1-6 alkyl group means that a hydrogen atom on a C alkyl group is substituted with a hydroxyl group or CON (R F ) R.
- a substituent selected from the group consisting of preferably 1 to 4, more preferably 1 to 3.
- a 5- to 6-membered heterocyclo which may be formed together with a nitrogen atom to which it is bonded, and may further include, as a ring-constituting atom, an oxygen atom, a sulfur atom, and a nitrogen nuclear atom.
- An alkyl group (the heterocycloalkyl group may be substituted with a hydroxyl group,
- C alkyl group optionally substituted with a hydroxyl group means at least one (preferably 1 to 4).
- hydroxymethyl group For example, hydroxymethyl group, hydroxyethyl group, 2-hydroxy 1,1-dimethylethyl group, 1,3 dihydroxy-2 methylpropane-2-yl group, 1,3 dihydroxy 2-hydroxymethylpropane 2- Group.
- “Pharmaceutically acceptable salt” is a salt with an alkali metal, an alkaline earth metal, ammonium, an alkyl ammonium, a mineral acid or an organic acid, For example, sodium salt, potassium salt, calcium salt, ammonium salt, aluminum salt, triethyl ammonium salt, acetate, propionate, butyrate, formate, trifluoroacetate, maleic acid Salt, tartrate, citrate, stearate, succinate, ethyl succinate, ratatobionate, darconate, darcoheptonate, benzoate, methanesulfonate, ethanesulfonate, 2 —Hydroxyethane sulfonate, benzene sulfonate, paratoluene sulfonate, lauryl sulfate, malate, aspartate, glutamate, adipate, salt with cysteine, salt with N-acetyl cysteine
- “Hydrate” is a pharmaceutically acceptable hydrate of the compound of the present invention or a salt thereof.
- the compound of the present invention or a salt thereof may be exposed to air or recrystallized to absorb moisture and form adsorbed water, or may become a hydrate.
- the hydrate in the present invention includes such hydrates.
- the compounds of the present invention may have chiral centers, they exist in various diastereomeric or enantiomeric forms. Some of the compounds of the present invention also exist as keto-enol tautomers, for example. In addition, some of the compounds of the present invention also exist as geometrically different bodies (E-form, Z-form). Accordingly, the compounds of the present invention include all the individual isomers above as well as mixtures thereof.
- the preferred U and group of Z are a methyl group, an ethyl group, a propyl group, and an isopropyl group, and more preferably an isopropyl group.
- R 1 is preferably a hydrogen atom or a methyl group
- R 2 and R 3 are preferably hydrogen atoms.
- the substitution position is preferably a meta position with respect to Y—Q! /.
- a preferred embodiment of Y is a C alkylene group, a C alkylene group, or —O— (CH 3).
- Y is preferably a C alkylene group or C alkyl
- R A and R B are the same or different and are selected from a hydrogen atom or a group power of “hydroxyl group and —CONH” 1 to 4 Pieces (more preferably 1-3)
- R A is —CONHR D (R D is a V, C alkyl group substituted with a hydroxyl group), and R B is a hydrogen atom. It is the case.
- a preferred embodiment when Q is —CONHR e is a C alkyl group substituted with 1 to 4 (more preferably 1 to 3) hydroxyl groups, or —CONH, —CONHC
- a C 2 alkyl cycloalkyl group substituted with any one of a CO heterocycloalkyl group (which may be substituted with a C alkyl group optionally substituted with a hydroxyl group).
- the carbon atom bonded to is tertiary.
- —Y—Q is more preferably —Y—CO—NH—C
- Y is propylene or pro
- Q is a hydroxyl group, OC alkylphenyl (for example,
- pyrazole derivative 1A can be synthesized in accordance with International Publication WO2004Z018491.
- the 5-thioglucose derivative 2A can be synthesized according to International Publication WO2004Z014931.
- 8-D-darcoside derivative 3A can be selectively produced from pyrazole derivative 1A and 5-thiodarcose derivative 2A.
- phosphines necessary as reagents in this reaction include triphenylphosphine, tri-n-butylphosphine, tri-butylphosphine, tristolylphosphine, diphenyl-2-pyridylphosphine, and the like. Of these, triphenylphosphine and diphenyl-1-pyridylphosphine are preferred, and triphenylphosphine is more preferred.
- azo reagents include jetyl azodicarboxylate, diisopropyl azodicarboxyloxy tert-butyl azodicarboxylate, 1,1'-azobis (N, N-dimethylformamide), 1,1 ′-(azodicarbol) dipiperidine can be used.
- jetylazodicarboxylate (DEAD) and diisopropylazodicarboxylate are preferable.
- Solvents used in this reaction are tetrahydrofuran, dioxane, toluene, methylene chloride, chloroform, acetonitrile, ethyl acetate, dimethyl sulfoxide, N, N dimethylformamide, etc., preferably tetrahydrofuran and toluene.
- the reaction temperature is preferably from 20 ° C to room temperature, more preferably from 5 ° C to + 5 ° C.
- the benzyl group can be removed by catalytic hydrogenation of the compound 3A obtained above using a catalyst such as palladium activated carbon, palladium hydroxide, or platinum-palladium activated carbon in a hydrogen atmosphere.
- a catalyst such as palladium activated carbon, palladium hydroxide, or platinum-palladium activated carbon in a hydrogen atmosphere.
- palladium hydroxide is preferred as the catalyst.
- the solvent used in this reaction include methanol, ethanol, isopropanol, ethyl acetate, acetic acid and the like.
- the reaction temperature is from room temperature to reflux temperature, with room temperature being preferred.
- the compound 5A can be obtained by converting the hydroxyl group of the compound 4A obtained above into an amino group.
- compound 4A was used in the presence of a base such as triethylamine, Nethyl N, N-diisopropylamine, pyridine, potassium carbonate, calcium carbonate, cesium carbonate, etc. Then, a leaving group is introduced.
- a base such as triethylamine, Nethyl N, N-diisopropylamine, pyridine, potassium carbonate, calcium carbonate, cesium carbonate, etc.
- a leaving group is introduced.
- the solvent used for this reaction include chloroform, dichloromethane, jetyl ether, tetrahydrofuran, dimethoxyethane, and ethyl acetate.
- the reaction temperature is from 0 ° C to room temperature.
- the azide group is converted to an amino group by catalytic hydrogenation similar to the above-mentioned debenzylation conditions to obtain compound 5A.
- compound 5B in which Y is a single bond is more preferably synthesized by the following method.
- the symbols are as defined above.
- pyrazole derivative 1B can be synthesized according to International Publication WO2004Z019958.
- compound 3B can be synthesized from pyrazole derivative 1B by the Mitsunobu reaction shown in step 1 of production method 1.
- the -tro group can be reduced by catalytic hydrogenation of Compound 3B under a hydrogen atmosphere using a catalyst such as palladium activated carbon, palladium hydroxide, or platinum oxide.
- a catalyst such as palladium activated carbon, palladium hydroxide, or platinum oxide.
- platinum-based catalysts such as platinum oxide are preferred.
- the solvent used in this reaction include methanol, ethanol, isopropanol, ethyl acetate, and acetic acid.
- the reaction temperature is from room temperature to reflux temperature, but room temperature is preferred.
- Compound 5A can be derivatized to compound 7A using guadinating reagent 6A.
- the solvent used in this reaction include tetrahydrofuran, N, N-dimethylformamide, methanol, ethanol, isopropanol, ethyl acetate, and toluene.
- the reaction temperature is from room temperature to reflux temperature.
- the preferred catalyst at this time is palladium hydroxide.
- Q is —N (R A ) R B , and R A is —CONHR D , R B Is a hydrogen atom (wherein R D represents a C alkyl group which may be substituted with a hydrogen atom or a substituent selected from the group consisting of a hydroxyl group and a pyridyl group).
- Compound 5A is condensed with ammine R D NH using p-nitrophenyl chloroformate, triphosgene or N, N'-carbodiimidazole (CDI) in the presence of a suitable base.
- Suitable bases include triethylamine, Nethyl N, N diisopropylamine, pyridine, DBU, potassium carbonate, calcium carbonate, cesium carbonate and the like.
- examples of the solvent used include chloroform, dichloromethane, jetyl ether, tetrahydrofuran, N, N dimethylformamide, acetonitrile, and ethyl acetate.
- the reaction temperature is from 0 ° C to the reflux temperature.
- Q is —N (R A ) R B , and R A and R B are the same or different and are a hydrogen atom or a C alkyl group (the C alkyl group is , Hydroxyl group, C ONH and OC alkyl-Fluca are group forces that are substituted with selected substituents
- L1 represents a halogen atom, a leaving group such as MeSO 0-, and the other symbols are as defined above.
- Compound 9A is obtained by reacting compound 5A with R A L1 or R B L1 in the presence of a suitable base.
- suitable bases include triethylamine, N ethyl N, N disopropylamine, pyridine, DBU, potassium carbonate, calcium carbonate, cesium carbonate and the like.
- the solvent to be used include N, N dimethylformamide, acetonitrile, methanol, ethanol, chlorophenol, dichloromethane, jetinoreethenole, tetrahydrofuran and the like.
- the reaction temperature is room temperature and reflux temperature.
- Compound 9A can also be produced under the same conditions as for conversion from compound 4A to the amino group of production method 1.
- compound 4A was prepared using methanesulfuryl chloride in the presence of a base such as triethylamine, Nethyl N, N diisopropylamine, pyridine, potassium carbonate, calcium carbonate, cesium carbonate, etc. using p-toluenesulfuryl chloride. Introduce a leaving group.
- the solvent used for this reaction include black mouth form, dichloromethane, jetyl ether, tetrahydrofuran, dimethoxyethane, and ethyl acetate.
- the reaction temperature is from 0 ° C to room temperature.
- triethinoreamine, N ethinorei N, N diisopropylamine, pyridine, DBU Compound 9A is obtained by reacting with NHR A R B in the presence of a suitable base such as potassium carbonate, calcium carbonate, cesium carbonate or the like.
- a suitable base such as potassium carbonate, calcium carbonate, cesium carbonate or the like.
- the solvent used in this reaction include N, N-dimethylformamide, dimethyl sulfoxide, N-methylpyrrolidone, dichloromethane, jetyl ether, tetrahydrofuran, dimethoxyethane, and ethyl acetate.
- the reaction temperature is from room temperature to reflux temperature.
- Q is —CONHR e
- Y is a C alkylene group
- a compound having a C alkellene group can be synthesized by the following method.
- Y 1 represents a single bond or a C alkylene group, and other symbols are as defined above.
- pyrazole derivative 1C can be synthesized in accordance with International Publication WO2004Z014932.
- the compound 3C can be synthesized from the pyrazole derivative 1C by the Mitsunobu reaction shown in Step 1 of Production Method 1.
- Compound 11A can be synthesized by subjecting compound 3C and olefin acetic acid 10A to a Heck reaction in the presence of a palladium catalyst, a phosphine ligand, and an appropriate base.
- a palladium catalyst used at this time include palladium acetate, tetrakistriphenylphosphine palladium, dibenzylideneacetone palladium, bistriphenylphosphine palladium chloride, palladium activated carbon and the like.
- the phosphine ligand include triphenylphosphine tris (2-methylphenol) phosphine.
- triethylamine, N-ethyl-N, N-diisopropylamine, potassium carbonate, calcium carbonate, cesium carbonate, potassium t-butoxide and the like are used as the base.
- the solvent used in the reaction include acetonitrile, toluene, tetrahydrofuran and the like.
- the reaction temperature ranges from 0 ° C to the reflux temperature, but a microwave may be used.
- reaction temperature is 0 ° C to 60 ° C.
- Compound 12B can also be produced by condensing the olefin moiety of Compound 11A with catalytic amine addition shown in Step 2 of Production Method 1 and then condensing with ammine (R e NH 3).
- Q is —CONHR e
- Y is —O— (CH 2)
- a compound can be synthesized by the following method.
- Y 1 represents a C alkylene group
- L2 represents a leaving group such as a halogen atom, MeSO 2 O-, etc.
- pyrazole derivative 4B can be synthesized in accordance with steps 1 and 2 of production method 1.
- Compound 12C can be obtained by reacting pyrazole derivative 4B with compound 13A in the presence of a suitable base.
- a suitable base Sodium hydride, potassium carbonate, cesium carbonate, sodium carbonate, and n-butyllithium are preferred as suitable bases.
- Solvents used in this reaction include tetrahydrofuran, jetyl ether, N, N-dimethylformamide, Seton and DMSO are preferred.
- the temperature of this reaction is 0 ° C to 60 ° C.
- the compound of the present invention selectively inhibits SGLT1, suppresses absorption of sugar from the small intestine,
- IGT can be improved.
- the compound of the present invention can be used as an active ingredient of an SGLT1 inhibitor or a preventive or therapeutic agent for diabetes, diabetes-related diseases and diabetic complications.
- diabetes includes type 1 diabetes, type 2 diabetes, and other types of diabetes caused by a specific cause.
- diabetes-related disease refers to obesity, hyperinsulinemia, abnormal glucose metabolism, hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, abnormal lipid metabolism, hypertension, congestive heart failure Edema, hyperuricemia, gout and the like.
- diabetes complications are classified into acute complications and chronic complications.
- Acute complications include hyperglycemia (such as ketoacidosis), infection (such as skin, soft tissue, biliary system, respiratory system, urinary tract infection) and the like.
- hyperglycemia such as ketoacidosis
- infection such as skin, soft tissue, biliary system, respiratory system, urinary tract infection
- Chronic complications include microangiopathy (nephropathy, retinopathy), arteriosclerosis (eg, atherosclerosis, myocardial infarction, cerebral infarction, lower limb arterial occlusion), neuropathy (sensory nerve, Motor nerves, autonomic nerves, etc.) and foot gangrene.
- microangiopathy nephropathy, retinopathy
- arteriosclerosis eg, atherosclerosis, myocardial infarction, cerebral infarction, lower limb arterial occlusion
- neuropathy sensor nerve, Motor nerves, autonomic nerves, etc.
- foot gangrene foot gangrene.
- the major complications are diabetic retinopathy, diabetic nephropathy, diabetic neuropathy.
- the compound of the present invention can be administered as a medicine systemically or locally, orally or parenterally.
- the compound of the present invention is a therapeutic agent for diabetes and a diabetic complication having a different mechanism of action other than SGLT1 and SGLT2 activity inhibitors for the purpose of enhancing the action of the compound or reducing the dose of the compound. It can be used in combination with drugs (hereinafter abbreviated as concomitant drugs) such as therapeutic agents, antihyperlipidemic agents, antihypertensive agents, antiobesity agents, diuretics and antithrombotic agents.
- concomitant drugs drugs
- the administration time of the compound of the present invention and the concomitant drug is not limited, and these may be administered simultaneously to the administration subject or may be administered with a time difference.
- the compound of the present invention and the concomitant drug may be administered as two types of preparations containing each active ingredient, or may be administered as a single preparation containing both active ingredients.
- the dose of the concomitant drug can be appropriately selected based on the clinically used dose.
- the compounding ratio of the compound of the present invention and the concomitant drug is the subject of administration, administration route, target disease, symptoms, combination It can be appropriately selected depending on the combination.
- the concomitant drug may be used in an amount of 0.01 to LOO parts by weight per 1 part by weight of the compound of the present invention.
- diabetes therapeutic agents include insulin preparations (eg, animal insulin preparations extracted from sushi and swine spleen; human insulin preparations synthesized using E. coli or yeast and genetically engineered; Protamine insulin sub-insulin fragments or derivatives (eg, INS-1 etc.), oral insulin preparations), insulin resistance improvers (eg,
- Pioglitazone or its salt preferably hydrochloride
- rosiglitazone or its salt preferably maleate
- riboglitazone CS—Oi l
- Sipoglitazar TAK— 654
- Metaglidasen M XB- 1 0 2
- Naveglitazar LY—519818
- MX—6054 Nolaglitazone
- N—2344 Nolaglitazone
- T—131 AMG131
- PPAR y Ago-st PPAR y-antagost
- a-Darcosidase inhibitor eg, voglibose, carbolose, miglitol, emidalitate
- biguanide eg, phenformin, metformin, buformin or Their salts (eg, hydrochloride, fumarate, succinate)
- insulin secretagogues sulfururea
- an aldose reductase inhibitor eg, torres tart, epanolorestat, zenarestat, zoponolestat, minanorestat, fidarestat, CT-112
- Neurotrophic factor and its increasing drug eg, NGF, NT-3, BDNF, neurotrophin production 'secretion promoter
- nerve regeneration promoter eg, Y-128
- PKC inhibitor eg, ruboxis
- Taurine mesylate LY- 333531
- AGE inhibitors eg, ALT946, pimagedin, pyratoxatin, N-phenacyl thiazolium bromide (ALT766), ALT-711, EXO-226, pyridorin
- active oxygen scavengers eg, thiotate
- cerebral vasodilators eg, thiopride, mexiletine
- Antihyperlipidemic agents include, for example, statin compounds (eg, pravastatin, sympastatin, oral pastatin, atonolepastatin, flupastatin, itapastatin, rosbastatin, pitapastatin or salts thereof (eg, Sodium salts, calcium salts)), squalene synthase inhibitors (eg, TAK-475), fibrate compounds (eg, bezafibrate, clofibrate, simfibrate, clinofibrate), AC AT inhibitors (eg, abashimibe ( Avasimibe), Eflucimibe), anion exchange scab (eg, cholestyramine), probuconole, nicotinic acid drugs (eg, nicomol), niceritro Niceritrol), icosapentate ethyl, plant sterols (eg, soysterol, gamma-oryzanol), CETP inhibitors (eg,
- antihypertensive agent examples include angiotensin converting enzyme inhibitors (eg, captopril, enalapril, delapril), angiotensin II antagonists (eg, candesartan cilexetil, oral sultan, eprosartan, valsartan, telmisartan, Ilbesartan, tasosartan, azilsartan (TAK-536)), calcium antagonists (eg, iliapine, difedipine, amlodipine, efonidipine, dicanodipine), potassium channelnore openers (eg, lebucromacarim, L 27152, AL0671, NIP — 121), including clodin
- Anti-obesity agents include, for example, central anti-obesity agents (eg, dexfenfluramine, phenfluramine, phentermine, sibutramine, amphepramone, dexamphetamine, mazindol, phenolpropanolamine, clobenzolex).
- central anti-obesity agents eg, dexfenfluramine, phenfluramine, phentermine, sibutramine, amphepramone, dexamphetamine, mazindol, phenolpropanolamine, clobenzolex.
- MCH receptor antagonists eg, compounds described in W 006/035967, SB—568849; SNAP—7941, T 226296); Neuropeptide antagonists (eg, CP—422935); Cannapinoid receptor antagonists (Eg, Rimonabant (SR—141716), SR—147778); ghrelin antagonists; 1 1 ⁇ -hydroxysteroid dehydrogenase inhibitors (eg, BVT-3498, INCB1373 9)), spleen lipase inhibitors ( Eg, orlistat, ATL—962), DGAT—1 inhibitor, / 3 3 agonist (eg, AJ—9677, AZ40140), peptidic appetite suppressant (eg, leptin, CN TF (ciliary neurotrophic) factor)) , Cholecyst kyungagost (eg, Lynch tribute, FPL-15849), antifeedant (eg, P-57).
- Neuropeptide antagonists
- diuretics examples include xanthine derivatives (eg, sodium salicylate theopromine, calcium salicylate theopromine), thiazide preparations (eg, ethiazide, cyclopenthiazide, trichloromethiazide, hydrothiazide, hydroflumethiazide, benzilhydride.
- xanthine derivatives eg, sodium salicylate theopromine, calcium salicylate theopromine
- thiazide preparations eg, ethiazide, cyclopenthiazide, trichloromethiazide, hydrothiazide, hydroflumethiazide, benzilhydride.
- Mouth-mouth mouth thiazide, penflutide, polythiazide, methycrothiazide), anti-aldosterone preparation eg, spironolatatone, triamterene
- carbonic anhydrase inhibitor eg, acetazolamide
- chlorobenzenesulfonamide preparation eg, chlorthalidone, mefluside, indapamide
- Examples include fazosemide, isosorbide, ethacrynic acid, pyrethradide, bumetide, and furosemide.
- Antithrombotic agents include, for example, heparin (eg, heparin sodium, heparin calcium, dalteparin sodium, AVE-5026), sulfarin (eg, sulfarin potassium), antithrombin Drugs (eg, argatroban, xymeragatran (Dimigatran), Dabigatran (Odiparcil, Lepirudin, bival irudin, Desirudin, ART—123, Idraparinux, SR—123781, AZD—0837, MCC—977, TGN— 255, TGN—167, RWJ—58436, LB—30870, MPC—09 20, Pegmusirudin ⁇ Org—426751, etc., thrombolytic drugs (eg, urokina se, tisokinase, anoleplase,reteplase) (natepla se), monteplase, pamiteplase, etc.), platelet
- a pharmaceutically acceptable carrier can be blended.
- examples of such carriers include common excipients, bulking agents, binders, disintegrants, coating agents, dragees, pH adjusters, solubilizers or aqueous or non-aqueous solvents. Tablets, pills, capsules, granules, powders, powders, solutions, emulsions, suspensions, injections and the like can be prepared from the compound of the present invention and these carriers.
- the solubility of the compound of the present invention can also be improved by inclusion in a, ⁇ or ⁇ -cyclodextrin, methylicyclodextrin, or the like.
- the dose of the compound of the present invention varies depending on the disease, symptom, body weight, age, sex, route of administration, etc. S, for adults, 0.1 to 1 per person: LOOOmg / kg body weight Yes, 0.1 to 200 mg Zkg body weight is preferred 0.1 to 10 mg Zkg body weight is more preferred. This can be administered once to several times a day.
- Example 2 4- [4- (2 Hydroxyethoxy) 2-methylbenzyl] -5-isopropyl 1H 1 Pyrazole 3-yl 2, 3, 4, 6-Tetra 1 O acetyl 3 synthesized in (1) 13-D-Dalcobilanoside (8. 38 g, 13.2 mmol) and triethylamine (2.8 mL, 20 mmol) in chlorosulfur chloride (1.2 mL, 13 mmol) under ice-cooling were added to a chloroform solution (82 mL). ) And stirred at room temperature for 30 minutes. A 0.5N aqueous hydrochloric acid solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the organic layer was washed with a saturated aqueous sodium bicarbonate solution and saturated brine, and dried over magnesium sulfate. After the desiccant was filtered off, the solvent was distilled off under reduced pressure. The residue was dissolved in N, N-dimethylformamide, sodium azide was added, and the mixture was stirred at 100 ° C. for 3 hours. Water was added to the reaction mixture, and the mixture was extracted twice with ethyl acetate. The organic layer was washed 3 times with saturated brine and dried over magnesium sulfate.
- Example 3 4- [4- (2 aminoethoxy) 2-methylbenzyl] -5-isopropyl 1H synthesized pyrazole 1-3-yl 2, 3, 4, 6-tetra 1 O synthesized in (2) Acetyl 5 -thio j8- D-Dalcoviranoside (0.145 g, 0.228 mmol) in chloroform (1.5 mL) solution with triethylmine (0.043 mL, 0.308 mmol), chloroformate 4 ditrophenol ( 0.053 g, 0.262 mmol) was added, and the mixture was stirred at room temperature for 1 hour.
- the reaction mixture was concentrated under reduced pressure, the residue was dissolved in methanol (l mL), sodium methoxide (25% methanol solution) was added, and the mixture was stirred at room temperature for 2 hr. After dry ice was added to the reaction solution for neutralization, the solvent was distilled off under reduced pressure.
- Example 13 (3 ⁇ ) — 4— [4— (Isopropyl 1- 3 — [(2,3,4,6—Tetra 1 ⁇ Acetinore 1 5 Thio 13-D-Darcopyranosyl) oxy) ] 1 ⁇ Pyrazol® 4-yl ⁇ methyl) phenol] buta-3-enic acid (0.728 g, 1.13 mmol) in methanol (5 mL) was added 10% palladium-activated carbon (70 mg) and hydrogen Stirred in the atmosphere. The reaction mixture was filtered through celite, and the filtrate was concentrated to give the title compound (0.670 g, 91%).
- the drug (the compound of the present invention) is mixed with lactose monohydrate, crystalline cellulose, carboxymethyl cellulose-calcium and hydroxypropylcellulose, and this mixture is pulverized with a pulverizer.
- the pulverized mixture is mixed with a stirring granulator for 1 minute, and then granulated with water for 4-8 minutes.
- the resulting granulate is dried at 70 ° C for 40 minutes.
- the granulated dry powder after sieving and magnesium stearate are mixed at 30 rpm for 3 minutes using a V-type mixer.
- the granules for tableting obtained using a mouth-tally tablet press are compression-molded and tableted.
- a human SGLT1 sequence (NM-000343) was amplified from human small intestine-derived mRNA after reverse transcription and introduced into pCMV-tag5A (Stratagene).
- a human SGLT2 sequence (NM-003 041) was prepared from human kidney-derived mRNA by the same method and introduced into pcDNA3.1 + hygro (Invitrogen). The sequence ability of each clone was confirmed to match the reported sequence.
- SGLT expressing cells are 50 Resistant strains were selected by culturing in the presence of dieneticin (SGLT1) or hygromycin B (SGLT2) at a concentration of 0 ⁇ gZmL, and the glucose uptake specific activity was obtained using the system shown below as an index.
- SGLT1 dieneticin
- SGLT2 hygromycin B
- Cells stably expressing human SGLT1 or human SGLT2 were used in a sodium-dependent glucose uptake activity inhibition test.
- Pretreatment buffer 140 mM choline chloride, 2 mM KC1, ImM CaCl, ImM Mg
- test compound ["C] methyl ⁇ -D-dalcopyranoside containing ⁇ -D-darcobilanoside (lmM), 145 mM NaCl, 2 mM KC1, ImM CaCl , ImM MgCl, lOmM HEPES / 5mM Tris, pH7.4
- the drug group was orally administered with a drug (lmgZkg) suspended in a 0.5% carboxymethylcellulose (CMC) aqueous solution and only a 0.5% CMC aqueous solution in the control group.
- CMC carboxymethylcellulose
- glucose solution (2 gZkg) was orally administered, and blood was collected at a total of 5 points before drug administration (Otime) and 0.25, 0.5, 1, 2 hours after oral administration.
- SGLT1 sodium-dependent glucose cotransporter expressed in the intestinal epithelium
- IGT abnormal glucose tolerance
Abstract
Disclosed is a novel pyrazolyl 5-thio-β-D-glucopyranoside compound which can inhibit the SGLT1 activity selectively and can prevent the absorption of glucose through the digestive tract, thereby controlling the IGT (impaired glucose tolerance). Specifically, disclosed is a pyrazolyl 5-thioglucoside compound represented by the formula (I), a pharmaceutically acceptable salt thereof, or a hydrate of the compound or the salt. [Chemical formula] wherein Z represents a C1-6 alkyl group; R1, R2 and R3 independently represent a hydrogen atom, a halogen atom, a C1-6 alkoxy group or a C1-6alkyl group; Y represents a single bond, a C1-6 alkylene group, a C2-6 alkenylene group or –O-(CH2)n- [wherein n represents an integer ranging from 1 to 4]; and Q represents a hydroxyl group, -OC1-4-alkylphenyl, -N(RA)RB or –CONHRC.
Description
明 細 書 Specification
ピラゾリル 5 _チォダルコシド化合物 Pyrazolyl 5_thiodarcoside compound
技術分野 Technical field
[0001] 本発明は、腎臓でのグルコース再吸収に関わるナトリウム依存性グルコース共輸送 体 1 (SGLT1)の阻害活性を有するピラゾリル 5—チォダルコシドィ匕合物に関する。 背景技術 [0001] The present invention relates to a pyrazolyl 5-thiodarcoside compound having an inhibitory activity on sodium-dependent glucose cotransporter 1 (SGLT1) involved in glucose reabsorption in the kidney. Background art
[0002] 糖尿病に罹患すると、空腹時の血糖値は 126mgZdL以上を示す。また、空腹時 の血糖値が正常であっても、食事の後に 140〜200mg/dLという高い血糖値を示 す場合には、耐糖能異常(以下、 IGT (impaired glucose tolerance)という。 )と診断さ れる。 IGTから糖尿病の発症を遅らせることは、心血管障害のリスクを低減させると考 えられ、それを示す幾つかの知見が得られている。例えば、 1997年に中国で行われ た Da Qing IGT and Diabetes Studyでは、ダイエットや運動を行うことで IGTから 2型 糖尿病への移行を有意に抑制したと報告されている (非特許文献 1参照)。また、薬剤 治療が有効な例として、糖の加水分解酵素を阻害し、小腸力 の糖の吸収を遅延さ せる a—ダルコシダーゼ阻害剤ァカルボースを投与すると、 IGTから 2型糖尿病への 移行を抑制し、さらに高血圧の発症も有意に抑制することが報告されている (非特許 文献 2参照)。 [0002] When suffering from diabetes, the fasting blood glucose level is 126 mgZdL or more. In addition, even if the fasting blood glucose level is normal, if it shows a high blood glucose level of 140 to 200 mg / dL after a meal, it is diagnosed as impaired glucose tolerance (hereinafter referred to as IGT (impaired glucose tolerance)). It is done. Delaying the onset of diabetes from IGT is thought to reduce the risk of cardiovascular disorders, and several findings have been obtained. For example, the Da Qing IGT and Diabetes Study conducted in China in 1997 reported that diet and exercise significantly suppressed the transition from IGT to type 2 diabetes (see Non-Patent Document 1). . In addition, as an effective example of drug treatment, administration of the a-darcosidase inhibitor acarbose, which inhibits sugar hydrolase and delays the absorption of sugar in the small intestine, suppresses the transition from IGT to type 2 diabetes. Furthermore, it has been reported that the onset of hypertension is also significantly suppressed (see Non-Patent Document 2).
[0003] この様なことから、糖尿病の発症を抑えるには、食事療法、運動及び薬物療法によ つて IGTをコント口一ルすることが重要である。 [0003] For these reasons, in order to suppress the onset of diabetes, it is important to control IGT by diet, exercise and drug therapy.
[0004] 哺乳動物の小腸上皮には高い頻度でナトリウム依存性グルコース共輸送体 1 (SG LT1)が発現している。この SGLT1は小腸において、ナトリウムに依存し、ダルコ—ス 又はガラクトースの能動輸送を司っていることが知られている。そこで、食事由来のグ ルコ—ス吸収を抑制し、 IGTの予防または治療を行うというコンセプトに基づき、 SGL T1活性を阻害するグリコシド化合物が報告されている (特許文献 1〜6参照)。 [0004] Sodium-dependent glucose cotransporter 1 (SG LT1) is frequently expressed in the small intestinal epithelium of mammals. SGLT1 is known to be dependent on sodium in the small intestine, and is responsible for active transport of dalcose or galactose. Thus, glycoside compounds that inhibit SGL T1 activity have been reported based on the concept of suppressing dietary glucose absorption and preventing or treating IGT (see Patent Documents 1 to 6).
[0005] また、腎臓には高頻度にナトリウム依存性ダルコース共輸送体 2 (SGLT2)が発現 しており、糸球体でー且濾過されたグルコースは SGLT2を介して再吸収される(非 特許文献 3参照)。そして、 SGLT2阻害剤は、尿への糖排泄を促進し、血糖低下作
用を招来するので、新たな糖尿病治療薬の標的分子と考えられるようになった (非特 許文献 4参照)。このような背景から、 SGLT2阻害剤が研究されヘテロァリ—ル 5— チォダルコシド誘導体が提供されて ヽる (特許文献 7参照)。 [0005] In addition, sodium-dependent dalcose cotransporter 2 (SGLT2) is frequently expressed in the kidney, and glucose that has been glomerularly filtered is reabsorbed via SGLT2 (Non-Patent Documents). 3). SGLT2 inhibitors promote glucose excretion in the urine and reduce blood sugar. As a result, it has come to be considered as a target molecule for new antidiabetic drugs (see Non-Patent Document 4). Against this background, SGLT2 inhibitors have been studied and heteroaryl 5-thiodarcoside derivatives have been provided (see Patent Document 7).
[0006] し力しながら、従来 SGLT1阻害活性を有するチォダルコシド誘導体は知られてい なかった。 [0006] However, a thiodarcoside derivative having SGLT1 inhibitory activity has not been known.
特許文献 1:国際公開第 WO2002Z098893号パンフレット Patent Document 1: International Publication No. WO2002Z098893 Pamphlet
特許文献 2:国際公開第 WO2004Z014932号パンフレット Patent Document 2: International Publication No. WO2004Z014932 Pamphlet
特許文献 3 :国際公開第 WO2004Z018491号パンフレット Patent Document 3: International Publication No. WO2004Z018491 Pamphlet
特許文献 4:国際公開第 WO2004Z019958号パンフレット Patent Document 4: International Publication No. WO2004Z019958 Pamphlet
特許文献 5 :国際公開第 WO2005Z121161号パンフレット Patent Document 5: International Publication No. WO2005Z121161 Pamphlet
特許文献 6 :国際公開第 WO2004Z050122号パンフレット Patent Document 6: International Publication No. WO2004Z050122 Pamphlet
特許文献 7:国際公開第 WO2004Z089967号パンフレット Patent Document 7: International Publication No. WO2004Z089967 Pamphlet
非特許文献 1 : Pan XR, et al. Diabets Care,第 20卷, 534頁, 1997年 Non-Patent Document 1: Pan XR, et al. Diabets Care, VIII, 534, 1997
非特許文献 2 : J.-し Chiasson, et al. Lancent,第 359卷, 2072頁, 2002年 Non-Patent Document 2: J.-Shi Chiasson, et al. Lancent, 359, 2072, 2002
非特許文献 3 : E. M. Wright, Am. J. Physiol. Renal. Physiol. ,第 280卷, F10項, 2001 年 Non-Patent Document 3: E. M. Wright, Am. J. Physiol. Renal. Physiol., 280, F10, 2001
非特許文献 4: G. Toggenburger, et al. Biochem. Biophys. Acta.,第 688卷, 557項, 1 982年 Non-Patent Document 4: G. Toggenburger, et al. Biochem. Biophys. Acta., 688, 557, 1 982
発明の開示 Disclosure of the invention
発明が解決しょうとする課題 Problems to be solved by the invention
[0007] 本発明は、選択的に SGLT1活性を阻害し、消化管からのグルコース吸収を抑制 することで、 IGTをコントロールする新規なピラゾリル 5—チォー 13 D—ダルコビラ ノシドィ匕合物を提供することを課題とする。 [0007] The present invention provides a novel pyrazolyl 5-thio-13D-darcoviranoside compound that controls IGT by selectively inhibiting SGLT1 activity and suppressing glucose absorption from the gastrointestinal tract. Is an issue.
課題を解決するための手段 Means for solving the problem
[0008] 本発明者らは前記課題を解決するために鋭意研究した結果、ある種のピラゾール 誘導体を 5 チォダルコシルイ匕した化合物が、優れた SGLT1活性阻害作用を有す ることを見出し、本発明を完成するに至った。 [0008] As a result of diligent research to solve the above-mentioned problems, the present inventors have found that a compound in which a certain pyrazole derivative is converted to 5-thiodalcosyl has an excellent SGLT1 activity inhibitory action. It came to be completed.
[0009] 以下に、本発明のピラゾリル 5 チォダルコシド誘導体 (以下、「本発明化合物」と
いう)の態様を述べる。 [0009] Hereinafter, the pyrazolyl 5 thiodarcoside derivative of the present invention (hereinafter referred to as "the compound of the present invention") )) Is described.
( 1 ) 本発明の態様(1 )は、下記式 (I)で表されるピラゾリル 5 チォダルコシドィ匕合 物若しくはその製薬学的に許容される塩又はそれらの水和物である。 (1) The embodiment (1) of the present invention is a pyrazolyl 5-thiodarcoside compound represented by the following formula (I), a pharmaceutically acceptable salt thereof, or a hydrate thereof.
[0010] [化 1] [0010] [Chemical 1]

( I ) (I)
[001 1] 式中、 Zは、 C アルキル基であり、 [001 1] In the formula, Z is a C alkyl group,
アルコキシ基
 Alkoxy group
Alkoxy group
または C アルキル基であり、 Or a C alkyl group,
1-6 1-6
Yは単結合、 C アルキレン基、 C ァルケ-レン基または— O— (CH ) n— (nは 1 Y is a single bond, a C alkylene group, a C alkylene group or —O— (CH) n— (n is 1
1-6 2-6 2 1-6 2-6 2
から 4の整数を示す)であり、 Represents an integer from 4 to 4)
Qは水酸基、 - OC アルキルフエ-ル、—N(RA)RBまたは CONHRCである、 Q is a hydroxyl group, -OC alkyl file, -N (R A ) R B or CONHRC,
1-4 1-4
但し、 Qが水酸基、 - OC アルキルフエ-ルまたは— NHのとき、 Yは単結合で However, when Q is a hydroxyl group, -OC alkylphenol or -NH, Y is a single bond.
1-4 2 1-4 2
はない、 No,
RA及び RBは同一または異なって、水素原子、—CONHRD (RDは水素原子ま たは 、水酸基及びピリジル基からなる群から選択される置換基で置換されてもよ!ヽ C ァ R A and R B may be the same or different and may be substituted with a hydrogen atom, —CONHR D (R D is a hydrogen atom or a substituent selected from the group consisting of a hydroxyl group and a pyridyl group!
1 - 6 ルキル基を示す)、 C (二 NH) NHRE (REは水素原子、または— CO C アルキル 1-6 alkyl), C (2 NH) NHR E (R E is a hydrogen atom, or —CO C alkyl
2 1-4 フエニルである)、あるいは、 C アルキル基 (該 C アルキル基は 、水酸基、 CO 2 1-4 phenyl) or a C alkyl group (the C alkyl group is a hydroxyl group, CO
1-6 1-6 1-6 1-6
NH及び OC アルキルフエニルからなる群から選択される置 換基で置換されて Substituted with a substituent selected from the group consisting of NH and OC alkylphenyl.
2 1-4 2 1-4
もよい)であり、 Is good)
は水酸基及び— CON (RF) 力もなる群より選択される置換基で置換された C Is C substituted with a substituent selected from the group consisting of a hydroxyl group and a —CON (R F ) force
1- アルキル基であり、 1-alkyl group,
6 6
RF及び Rは、同一または異なって、水素原子または C アルキル基である力 ある
いは RF及び Rが結合している窒素原子と一緒になつて、さらに環構成原子として、 酸素原子、硫黄原子及び窒素原子から選択されるへテロ原子を含んでも良い 5〜6 員のへテロシクロアルキル基 (該ヘテロシクロアルキル基は水酸基で置換されても良R F and R are the same or different and may be a hydrogen atom or a C alkyl group There are R F and R a together with the nitrogen atom to which they are attached connexion, a further ring-constituting atom, an oxygen atom, a sulfur atom and to the to be good 5-6 membered comprise hetero atoms selected from nitrogen atom Telocycloalkyl group (the heterocycloalkyl group may be substituted with a hydroxyl group)
V、C アルキル基で置換されてもょ 、)を形成してもよ!/、。 V, C may be substituted with alkyl groups, may form)! / ,.
1-6 1-6
なお、ピラゾール環の点線は、環構成窒素原子に水素原子が結合する位置によつ て、互変異性体が存在することを示しているが、下記に示す具体的化合物では、便 宜上、どちらか一方の異性体のみを示す。 The dotted line of the pyrazole ring indicates that a tautomer exists depending on the position where the hydrogen atom is bonded to the ring nitrogen atom. For convenience, in the specific compound shown below, Only one of the isomers is shown.
(2) 本発明の他の態様は、 Yが C アルキレン基、 C ァルケ-レン基または—O— (2) In another embodiment of the present invention, Y is a C alkylene group, a C alkylene group or —O—.
1-6 2-6 1-6 2-6
(CH ) n—(nは 2から 4の整数である)である、態様(1)記載のピラゾリル 5 チォグ Pyrazolyl 5 thog according to embodiment (1), wherein (CH 2) n— (n is an integer from 2 to 4)
2 2
ルコシド化合物若しくはその製薬学的に許容される塩又はそれらの水和物である。It is a lucoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof.
(3) 本発明の他の態様は、 Qがー N(RA)RB (RA及び RBは態様(1)で定義したとおり である)である、態様(1)または(2)に記載のピラゾリル 5 チォダルコシドィ匕合物若 しくはその製薬学的に許容される塩又はそれらの水和物である。 (3) Another embodiment of the present invention is the embodiment (1) or (2), wherein Q is -N (R A ) R B (R A and R B are as defined in embodiment (1)). Or a pharmaceutically acceptable salt thereof or a hydrate thereof.
(4) 本発明の他の態様は、 RAが— CONHRD (RDは水酸基及びピリジル基力もなる 群から選択される置換基で置換されてもょ ヽ C アルキル基である)であり、 RBが水 (4) In another embodiment of the present invention, R A is —CONHR D (wherein R D is a C alkyl group substituted with a substituent selected from the group consisting of a hydroxyl group and a pyridyl group), R B is water
1-6 1-6
素原子である、態様(3)に記載のピラゾリル 5—チォダルコシドィ匕合物若しくはその 製薬学的に許容される塩又はそれらの水和物である。 A pyrazolyl 5-thiodarcoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof according to embodiment (3), which is a primary atom.
(5) 本発明の他の態様は、 RAが— C ( = NH) NHRE(REは水素原子、または— CO (5) In another embodiment of the present invention, R A is —C (═NH) NHR E (R E is a hydrogen atom, or —CO
2 2
C アルキルフエニルである)であり、 RBが水素原子である、態様(3)に記載のピラゾC. is an alkylphenyl), and R B is a hydrogen atom.
1-4 1-4
リル 5—チォダルコシド化合物若しくはその製薬学的に許容される塩又はそれらの 水和物である。 Ryl 5-thiodarcoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof.
(6) 本発明の他の態様は、 RA及び RBが同一または異なって、水素原子、または C (6) In another embodiment of the present invention, R A and R B are the same or different, and a hydrogen atom or C
1- アルキル基(該 C アルキル基は、水酸基、 CONH及び OC アルキルフエ二 1-alkyl group (the C alkyl group is a hydroxyl group, CONH and OC alkylphenol)
6 1-6 2 1-4 6 1-6 2 1-4
ルカもなる群力も選択される置換基で置換されてもよ!、)である態様 (3)に記載のビラ ゾリル 5—チォダルコシド化合物若しくはその製薬学的に許容される塩又はそれら の水和物である。 Luca may also be substituted with a selected substituent! ) Is a virazolyl 5-thiodarcoside compound according to embodiment (3), or a pharmaceutically acceptable salt thereof, or a hydrate thereof.
(7) 本発明の他の態様は、 Qがー CONHRe (Reは態様(1)で定義したとおりである )である、態様( 1)または(2)に記載のピラゾリル 5 -チォダルコシドィ匕合物若しくは
その製薬学的に許容される塩又はそれらの水和物である。 (7) Another embodiment of the present invention is a pyrazolyl 5-thiodarcosidoxy compound according to embodiment (1) or (2), wherein Q is -CONHR e (R e is as defined in embodiment (1)). Compound or The pharmaceutically acceptable salt or hydrate thereof.
(8) 本発明の他の態様は、 REが水酸基及び— CONHカゝらなる群より選択される 1 (8) In another embodiment of the present invention, R E is selected from the group consisting of a hydroxyl group and —CONH key 1
2 2
〜4個の置換基で置換された C アルキル基である力、または、 CO ピペラジノ( Force that is a C alkyl group substituted with ~ 4 substituents, or CO piperazino (
1-6 1-6
該ピペラジノは、水酸基で置換されても良い C アルキル基で置換されてもよい)で The piperazino may be substituted with a C alkyl group optionally substituted with a hydroxyl group)
1-6 1-6
置換された C アルキル基である(ここで、 RGが— NH—に結合する各 C アルキル A substituted C alkyl group, where R G is each C alkyl bonded to —NH—
1-6 1-6 基の炭素原子は 3級である)、態様 (7)記載のピラゾリル 5—チォダルコシド化合物 若しくはその製薬学的に許容される塩又はそれらの水和物である。 1-6 The carbon atom of 1-6 group is tertiary), and the pyrazolyl 5-thiodarcoside compound according to embodiment (7) or a pharmaceutically acceptable salt thereof or a hydrate thereof.
(9) 本発明の他の態様は、 Yは C アルキレン基又は C ァルケ-レン基である、態 (9) In another embodiment of the present invention, Y is a C alkylene group or a C alkene group.
1-6 2-6 1-6 2-6
様 (7)または (8)記載のピラゾリル 5—チォダルコシド化合物若しくはその製薬学的 に許容される塩又はそれらの水和物である。 (7) A pyrazolyl 5-thiodarcoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof according to (7) or (8).
(10) 本発明の他の態様は、 (10) Another aspect of the present invention is:
アルキル基であり、
 An alkyl group,
An alkyl group,
Yは、 O— (CH ) n- (nは 1から 4の整数である)であり、 Y is O— (CH 2) n- (n is an integer from 1 to 4);
2 2
Qは、水酸基、 OC アルキルフエニル、又は— N(RA)RBであり、 Q is a hydroxyl group, an OC alkylphenyl, or —N (R A ) R B ;
1-4 1-4
RA及び RBは、同一又は異なって、水素原子、 -CONHで置換された C アルキ R A and R B are the same or different and are each a hydrogen atom, C alkyl substituted with -CONH.
2 1-6 ル基、 CONHRD (RDは、水酸基で置換された C アルキル基である)、あるいは— 2 1-6 group, CONHR D (R D is a C alkyl group substituted with a hydroxyl group), or —
1-6 1-6
C (二 NH) NHRE (REは、水素原子または— CO C アルキルフエニルである)である C (2 NH) NHR E (R E is a hydrogen atom or —CO C alkylphenyl)
2 1-4 2 1-4
、態様(1)に記載のピラゾリル 5 チォダルコシドィ匕合物若しくはその製薬学的に許 容される塩又はそれらの水和物である。 The pyrazolyl 5 thiodarcoside compound or the pharmaceutically acceptable salt thereof or the hydrate thereof according to the embodiment (1).
(11) 本発明の他の態様は、態様(1)〜(10)のいずれかに記載のピラゾリル 5— チォダルコシドィ匕合物若しくはその製薬学的に許容される塩又はそれらの水和物を 有効成分として含む、 SGLT1活性阻害剤である。 (11) In another embodiment of the present invention, the pyrazolyl 5-thiodarcoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof according to any one of embodiments (1) to (10) is effective. SGLT1 activity inhibitor contained as a component.
(12) 本発明の他の態様は、態様(1)〜(10)のいずれかに記載のピラゾリル 5— チォダルコシドィ匕合物若しくはその製薬学的に許容される塩又はそれらの水和物を 有効成分として含む、糖尿病の予防又は治療剤である。 (12) In another embodiment of the present invention, the pyrazolyl 5-thiodarcoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof according to any one of embodiments (1) to (10) is effective. It is a preventive or therapeutic agent for diabetes included as a component.
発明の効果 The invention's effect
本発明の化合物は、 SGLT1活性を選択的に阻害するので、糖尿病患者の治療だ けでなぐ食前血糖が正常域であっても食後高血糖がみられる、境界型 (糖尿病予
備群)の患者の治療もすることができ、これにより糖尿病への移行予防をすることがで きる。 Since the compound of the present invention selectively inhibits SGLT1 activity, borderline type (diabetic It is possible to treat patients in the group), thereby preventing the transition to diabetes.
発明を実施するための最良の形態 BEST MODE FOR CARRYING OUT THE INVENTION
[0014] 本発明において使用する用語を以下に定義する。 [0014] Terms used in the present invention are defined below.
[0015] 「C アルキル基」とは、炭素原子を 1—6個有する直鎖状又は分枝状のアルキル基 “C alkyl group” means a linear or branched alkyl group having 1 to 6 carbon atoms.
1-6 1-6
を意味する。例えば、メチル基、ェチル基、 n プロピル基、イソプロピル基、 n—ブチ ル基、イソブチル基、 tert ブチル基、 sec ブチル基、 n ペンチル基、 n—へキシ ル基が挙げられる。 Means. For example, a methyl group, an ethyl group, an n propyl group, an isopropyl group, an n-butyl group, an isobutyl group, a tert butyl group, a sec butyl group, an n pentyl group, and an n-hexyl group can be mentioned.
[0016] 「C アルコキシ基」とは、炭素原子を 1 6個有する直鎖状又は分枝状のアルコキ [0016] "C alkoxy group" means a straight-chain or branched alkoxy having 16 carbon atoms.
1-6 1-6
シ基を意味し、 C アルコキシ基が好ましい。 C アルコキシ基としては、例えば、メト Means a Si group, and a C alkoxy group is preferred. Examples of the C alkoxy group include meth
1-4 1-4 1-4 1-4
キシ基、エトキシ基、プロポキシ基、イソプロポキシ基、 n ブトキシ基、イソブトキシ基 Xoxy group, ethoxy group, propoxy group, isopropoxy group, n butoxy group, isobutoxy group
、 tert ブトキシ基が挙げられる。 And tert butoxy group.
[0017] 「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子又はヨウ素原子である。 The “halogen atom” is a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
[0018] 「一 OC アルキルフエ-ル」とは、 C アルコキシ基とフエ-ルが複合した形態を有 [0018] "One OC alkyl file" has a form in which a C alkoxy group and a file are combined.
1-4 1-4 1-4 1-4
する基である。例えば、ベンジルォキシ基、フエニルェチルォキシ基が挙げられる。 It is a group. Examples thereof include a benzyloxy group and a phenylethyloxy group.
[0019] 「C アルキレン基」とは、炭素数 1〜6個からなる、直鎖または分岐状の 2価の飽和 [0019] The "C alkylene group" is a linear or branched divalent saturated group having 1 to 6 carbon atoms.
1- 6 1-6
炭化水素である。例えば、メチレン、エチレン、トリメチレン、テトラメチレン、 2—メチル プロピレン、 2, 2—ジメチルプロピレン等が挙げられる。 It is a hydrocarbon. For example, methylene, ethylene, trimethylene, tetramethylene, 2-methylpropylene, 2,2-dimethylpropylene and the like can be mentioned.
[0020] 「C ァルケ-レン基」とは、二重結合を含む炭素数 2〜6個力もなる、直鎖または分 [0020] The "C alk-lene group" is a straight chain or branched chain having 2 to 6 carbon atoms including a double bond.
2- 6 2- 6
岐状の 2価の不飽和炭化水素である。例えば、エチレン、プロべ-レン、 2—プチ-レ ン等が挙げられ、中でもプロべ-レンが好ましい。 It is a branched divalent unsaturated hydrocarbon. For example, ethylene, probelene, 2-butylene and the like can be mentioned. Of these, probelene is preferable.
[0021] 「水酸基及びピリジル基からなる群から選択される置換基で置換されてもよい C ァ [0021] “C-a which may be substituted with a substituent selected from the group consisting of a hydroxyl group and a pyridyl group”
1-6 ルキル基」とは、 c アルキル基上の水素原子が、水酸基及びピリジル基から選択さ “1-6 alkyl group” means that the hydrogen atom on the c alkyl group is selected from a hydroxyl group and a pyridyl group.
1-6 1-6
れる一つ以上の基 (好ましくは 1〜3個)によって置換されてもよい C アルキル基を A C alkyl group optionally substituted by one or more groups (preferably 1 to 3)
1-6 1-6
示す。例えば、メチル基、ェチル基、イソプロピル基、ピリジン 3—ィルメチル基、ヒ ドロキシメチル基、ヒドロキシェチル基、 2—ヒドロキシー 1,1ージメチルェチル基、 1, 3 -ジヒドロキシ 2 ヒドロキシメチルプロパン— 2 ィル基が挙げられる。 Show. For example, methyl group, ethyl group, isopropyl group, pyridine 3-ylmethyl group, hydroxymethyl group, hydroxyethyl group, 2-hydroxy-1,1-dimethylethyl group, 1,3-dihydroxy-2 hydroxymethylpropane-2-yl group Can be mentioned.
[0022] 「水酸基、 -CONH及び OC アルキルフ -ルからなる群から選択される置換
基で置換されてもよい c アルキル基」とは、 C アルキル基上の水素原子が、水酸 [0022] "Substitution selected from the group consisting of hydroxyl group, -CONH and OC alkylfur" C alkyl group optionally substituted with a group means that a hydrogen atom on a C alkyl group is a hydroxyl group.
1-6 1-6 1-6 1-6
基、 CONH及び OC アルキルフエニルからなる群から選択される 1つ以上(好 One or more selected from the group consisting of the groups CONH and OC alkylphenyl (preferably
2 1-4 2 1-4
ましくは 1〜3個)の基によって置換されてもよい C アルキル基を示す。例えば、メチ C represents an alkyl group which may be preferably substituted by 1 to 3 groups. For example, Mechi
1-6 1-6
ル基、ェチル基、イソプロピル基、ベンジルォキシェチル基、ヒドロキシメチル基、ヒド ロキシェチル基、 2 ヒドロキシ 1,1ージメチルェチル基、 1, 3 ヒドロキシー2—メ チルプロパン 2—ィル基、力ルバモイルメチル基、力ルバモイルメチル基、 2—カル バモイルェチル基、 2—力ルバモイルー 2—メチルェチル基が挙げられる。 Group, ethyl group, isopropyl group, benzyloxycetyl group, hydroxymethyl group, hydroxyxetyl group, 2 hydroxy 1,1-dimethylethyl group, 1,3 hydroxy-2-methylpropane 2-yl group, strong rubamoylmethyl group, Examples thereof include a strong ruberamoylmethyl group, a 2-carbamoylethyl group, and a 2-force rubamoyl-2-methylethyl group.
[0023] 「水酸基及び— CON (RF)R力もなる群より選択された置換基で置換された C ァ [0023] "Hydroxyl and C (a) substituted with a substituent selected from the group consisting of CON (R F ) R force"
1-6 ルキル基」とは、 C アルキル基上の水素原子が、水酸基または CON (RF)Rから “1-6 alkyl group” means that a hydrogen atom on a C alkyl group is substituted with a hydroxyl group or CON (R F ) R.
1-6 1-6
なる群より選択される置換基 (好ましくは 1〜4個、より好ましくは 1〜3個)によって置 換されている C アルキル基を示す。例えば、ヒドロキシメチル基、ヒドロキシェチル基 A C alkyl group substituted by a substituent selected from the group consisting of (preferably 1 to 4, more preferably 1 to 3). For example, hydroxymethyl group, hydroxyethyl group
1-6 1-6
、 2 ヒドロキシ 1,1ージメチルェチル基、 1, 3 ヒドロキシー2 メチルプロパン 2—ィル基、力ルバモイルメチル基が挙げられる。 2 hydroxy 1,1-dimethylethyl group, 1,3-hydroxy-2-methylpropane 2-yl group, and rubamoylmethyl group.
[0024] 「結合している窒素原子と一緒になつて形成し、さらに環構成原子として、酸素原子 、硫黄原子及び窒素原子力 選択されるへテロ原子を含んでも良い 5〜6員のへテロ シクロアルキル基 (該ヘテロシクロアルキル基は水酸基で置換されてもょ 、C アルキ [0024] "A 5- to 6-membered heterocyclo, which may be formed together with a nitrogen atom to which it is bonded, and may further include, as a ring-constituting atom, an oxygen atom, a sulfur atom, and a nitrogen nuclear atom. An alkyl group (the heterocycloalkyl group may be substituted with a hydroxyl group,
1-6 ル基で置換されてもよい)」の例としては、 1—ピロリジ -ル基、ピペリジノ基、 1—ピぺ ラジニル基、モルホリノ基、チオモルホリノ基、 4ーメチルー 1ーピペラジ-ル基等が挙 げられる。 Examples of 1-pyrrolidyl group, piperidino group, 1-piperazinyl group, morpholino group, thiomorpholino group, 4- methyl-1-piperazyl group, etc. Are listed.
[0025] 「水酸基で置換されてもよい C アルキル基」とは、少なくとも 1個(好ましくは 1〜4 [0025] "C alkyl group optionally substituted with a hydroxyl group" means at least one (preferably 1 to 4).
1-6 1-6
個、より好ましくは 1〜3個)の水酸基によって置換されてもよい C アルキル基を意味 Or more preferably 1 to 3) C alkyl groups which may be substituted by hydroxyl groups
1-6 1-6
し、例えば、ヒドロキシメチル基、ヒドロキシェチル基、 2—ヒドロキシ 1,1ージメチル ェチル基、 1,3 ジヒドロキシー2 メチルプロパンー2—ィル基、 1,3 ジヒドロキシ 2—ヒドロキシメチルプロパン 2—ィル基が挙げられる。 For example, hydroxymethyl group, hydroxyethyl group, 2-hydroxy 1,1-dimethylethyl group, 1,3 dihydroxy-2 methylpropane-2-yl group, 1,3 dihydroxy 2-hydroxymethylpropane 2- Group.
[0026] 「製薬学的に許容される塩」とは、アルカリ金属類、アルカリ土類金属類、アンモニゥ ム、アルキルアンモ-ゥムなどとの塩、鉱酸又は有機酸との塩であり、例えば、ナトリウ ム塩、カリウム塩、カルシウム塩、アンモ-ゥム塩、アルミニウム塩、トリェチルアンモ- ゥム塩、酢酸塩、プロピオン酸塩、酪酸塩、ぎ酸塩、トリフルォロ酢酸塩、マレイン酸
塩、酒石酸塩、クェン酸塩、ステアリン酸塩、コハク酸塩、ェチルコハク酸塩、ラタトビ オン酸塩、ダルコン酸塩、ダルコヘプトン酸塩、安息香酸塩、メタンスルホン酸塩、ェ タンスルホン酸塩、 2—ヒドロキシエタンスルホン酸塩、ベンゼンスルホン酸塩、パラト ルエンスルホン酸塩、ラウリル硫酸塩、リンゴ酸塩、ァスパラギン酸塩、グルタミン酸塩 、アジピン酸塩、システィンとの塩、 N—ァセチルシスティンとの塩、塩酸塩、臭化水 素酸塩、リン酸塩、硫酸塩、よう化水素酸塩、ニコチン酸塩、シユウ酸塩、ピクリン酸塩 、チォシアン酸塩、ゥンデカン酸塩、アクリル酸ポリマーとの塩、カルボキシビュルポリ マーとの塩が挙げられる。 [0026] "Pharmaceutically acceptable salt" is a salt with an alkali metal, an alkaline earth metal, ammonium, an alkyl ammonium, a mineral acid or an organic acid, For example, sodium salt, potassium salt, calcium salt, ammonium salt, aluminum salt, triethyl ammonium salt, acetate, propionate, butyrate, formate, trifluoroacetate, maleic acid Salt, tartrate, citrate, stearate, succinate, ethyl succinate, ratatobionate, darconate, darcoheptonate, benzoate, methanesulfonate, ethanesulfonate, 2 —Hydroxyethane sulfonate, benzene sulfonate, paratoluene sulfonate, lauryl sulfate, malate, aspartate, glutamate, adipate, salt with cysteine, salt with N-acetyl cysteine , Hydrochloride, hydrobromide, phosphate, sulfate, hydroiodide, nicotinate, oxalate, picrate, thiocyanate, undecanoate, salt with acrylic polymer And salts with carboxybutyl polymers.
[0027] 「水和物」とは、本発明の化合物又はその塩の製薬学的に許容される水和物である 。本発明の化合物又はその塩は、大気にさらされ、あるいは再結晶することなどにより 、水分を吸収し、吸着水がつく場合や、水和物となる場合がある。本発明における水 和物には、そのような水和物も含まれる。 “Hydrate” is a pharmaceutically acceptable hydrate of the compound of the present invention or a salt thereof. The compound of the present invention or a salt thereof may be exposed to air or recrystallized to absorb moisture and form adsorbed water, or may become a hydrate. The hydrate in the present invention includes such hydrates.
[0028] 本発明の化合物はキラル中心を有することがあるので、種々なジァステレオマー形 又はェナンチォマー形として存在する。また、本発明の化合物の一部は、例えば、ケ ト—エノール互変異性体としても存在する。また、本発明の化合物の一部は、幾何異 性体 (E体、 Z体)としても存在する。したがって、本発明の化合物は、上記全ての個 々の異性体並びにこれらの混合物を包含する。 [0028] Since the compounds of the present invention may have chiral centers, they exist in various diastereomeric or enantiomeric forms. Some of the compounds of the present invention also exist as keto-enol tautomers, for example. In addition, some of the compounds of the present invention also exist as geometrically different bodies (E-form, Z-form). Accordingly, the compounds of the present invention include all the individual isomers above as well as mixtures thereof.
[0029] 本発明化合物の好ましい態様を以下にあげる。 [0029] Preferred embodiments of the compound of the present invention are listed below.
[0030] 式 (I)にお!/、て、 Zの好ま U、基は、メチル基、ェチル基、プロピル基、イソプロピル 基であり、より好ましくはイソプロピル基である。 [0030] In the formula (I), the preferred U and group of Z are a methyl group, an ethyl group, a propyl group, and an isopropyl group, and more preferably an isopropyl group.
[0031] ルキル基であり、より好ま
 [0031] Rukiru group, more preferred
[0031] Rukiru group, more preferred
しくは、水素原子、またはメチル基である。特に、 R1が水素原子またはメチル基であり 、 R2及び R3が水素原子であることが好ましい。この場合、 R1がメチル基であるときの置 換位置は一 Y— Qに対してメタ位であることが好まし!/、。 Or a hydrogen atom or a methyl group. In particular, R 1 is preferably a hydrogen atom or a methyl group, and R 2 and R 3 are preferably hydrogen atoms. In this case, when R 1 is a methyl group, the substitution position is preferably a meta position with respect to Y—Q! /.
[0032] Yの好ましい態様は C アルキレン基、 C ァルケ-レン基、あるいは— O— (CH ) [0032] A preferred embodiment of Y is a C alkylene group, a C alkylene group, or —O— (CH 3).
1-4 2-4 2 n—(n= 2または 3)である。 1-4 2-4 2 n— (n = 2 or 3).
[0033] Qが— NRA(RB)である場合の好ましい態様は、 Yが C アルキレン基又は C アル [0033] In the case where Q is —NR A (R B ), Y is preferably a C alkylene group or C alkyl
1-4 2-4 ケ-レン基であり、より好ましくは、 Yが C アルキレン基である。
[0034] Qがー NRA(RB)の好ましい態様は、(1) RA及び RBは同一または異なって、水素原 子、または"水酸基及び— CONHの群力 選択される 1〜4個(より好ましくは 1〜3 1-4 2-4 a kelen group, more preferably Y is a C alkylene group. [0034] Preferred embodiments of Q-NR A (R B ) are as follows: (1) R A and R B are the same or different and are selected from a hydrogen atom or a group power of “hydroxyl group and —CONH” 1 to 4 Pieces (more preferably 1-3)
2 2
個)の基で置換されてもよい C アルキル基"である場合、(2) RAが— C ( = NH) NH In the case of a C alkyl group which may be substituted with a group of ()), (2) R A is — C (= NH) NH
1-6 2 であり、 が水素原子である場合、(3) RAが— CONHRD (RDは水酸基で置換されて V、る C アルキル基である)であり、 RBは水素原子である場合である。 1-6 2 and is a hydrogen atom, (3) R A is —CONHR D (R D is a V, C alkyl group substituted with a hydroxyl group), and R B is a hydrogen atom. It is the case.
1-6 1-6
[0035] Qが— CONHReである場合の好ましい態様は、 1〜4個(より好ましくは 1〜3個)の 水酸基で置換された C アルキル基、または— CONH 、 -CONHC アルキル若 [0035] A preferred embodiment when Q is —CONHR e is a C alkyl group substituted with 1 to 4 (more preferably 1 to 3) hydroxyl groups, or —CONH, —CONHC
1-6 2 1-4 1-6 2 1-4
しくは CO ヘテロシクロアルキル基 (該ヘテロシクロアルキル基は、水酸基で置換 されてもよい C アルキル基で置換されてもよい)のいずれかで置換された C アルキ Or a C 2 alkyl cycloalkyl group substituted with any one of a CO heterocycloalkyl group (which may be substituted with a C alkyl group optionally substituted with a hydroxyl group).
1-6 1-6 ル基である。 CONH(RC)において、 RCに定義される C アルキル基は、 一 NH— 1-6 1-6 group. In CONH (R C ), the C alkyl group defined for R C is one NH—
1-6 1-6
に結合する炭素原子が 3級である場合が好まし 、。 Preferably, the carbon atom bonded to is tertiary.
[0036] 上記の化合物群において、より好ましい—Y—Qは、—Y—CO—NH— C アル In the above compound group, —Y—Q is more preferably —Y—CO—NH—C
1-6 キル (該アルキルの NHに結合する炭素原子が 3級であり、該アルキルは水酸基で置 換 されている)、 Y—CO— NH— C (CH ) —CONH 、 一 Y—CO— NH— C ( 1-6 Kill (The carbon atom bonded to NH of the alkyl is tertiary, and the alkyl is replaced with a hydroxyl group), Y—CO—NH—C (CH) —CONH, 1 Y—CO— NH— C (
3 2 2 3 2 2
CH ) — CONH— C アルキル (該アルキルは水酸基で置換されてもよい )、 一 Y CH) — CONH— C alkyl (the alkyl may be substituted with a hydroxyl group), Y
3 2 1-6 3 2 1-6
-CO-NH-C (CH ) CO ピペラジノ(該ピペラジノは、水酸基 で置換されて -CO-NH-C (CH) CO piperazino (this piperazino is substituted with a hydroxyl group
3 2 3 2
もよい C アルキル基で置換されてもよい)である。ここで、 Yは、プ ロピレン又はプロ It may be substituted with a C alkyl group). Where Y is propylene or pro
1-6 1-6
ベ-レン(一 CH = CH— CH—)である。 Belem (one CH = CH—CH—).
2 2
[0037] 以下に、本発明化合物 (I)の製造方法を例をあげて以下に詳細に説明するが、例 示されたものに特に限定されない。 [0037] Hereinafter, the production method of the compound (I) of the present invention will be described in detail with reference to examples, but the present invention is not particularly limited to the examples.
[0038] 製造法 1 [0038] Manufacturing method 1
本発明の式 (I)化合物において、 Qが水酸基、 OC アルキルフ ニル(例えば、 In the compound of formula (I) of the present invention, Q is a hydroxyl group, OC alkylphenyl (for example,
1-4 1-4
— OBn)又はアミノ基である化合物は以下の方法で合成できる。 — OBn) or an amino group can be synthesized by the following method.
[0039] ただし、 ルカノィル基又
 [0039] However, Lucanoyl group
[0039] However, Lucanoyl group
はベンゾィル基を示し、その他の記号は前記と同義である。 Represents a benzoyl group, and the other symbols are as defined above.
[0040] [化 2]
 [0040] [Chemical 2]
[0040] [Chemical 2]
[0041] ここに、ピラゾール誘導体 1Aは国際公開 WO2004Z018491号に準拠して合成 することができる。 5—チォグルコース誘導体 2Aは、国際公開 WO2004Z014931 号に準拠して合成することができる。 [0041] Here, pyrazole derivative 1A can be synthesized in accordance with International Publication WO2004Z018491. The 5-thioglucose derivative 2A can be synthesized according to International Publication WO2004Z014931.
(1)工程 1 (光延反応) (1) Process 1 (Mitsunobu reaction)
光延反応(国際公開 WO2004Z089966号)によって、ピラゾール誘導体 1Aと 5 -チォダルコース誘導体 2Aからピラゾリル 5—チォ— |8— D—ダルコシド誘導体 3 Aを選択的に製造することができる。この反応における試薬として必要なホスフィン類 の例としては、トリフエニルホスフィン、トリー n—ブチルホスフィン、トリー tーブチルホ スフイン、トリストリルホスフィンゃジフエ-ル一 2—ピリジルホスフィン等が挙げられる。 中でもトリフエ-ルホスフィン、ジフエ-ル一 2—ピリジルホスフィンが好ましぐトリフエ -ルホスフィンがより好まし 、。 By the Mitsunobu reaction (International Publication WO2004Z089966), pyrazolyl 5-thio- | 8-D-darcoside derivative 3A can be selectively produced from pyrazole derivative 1A and 5-thiodarcose derivative 2A. Examples of phosphines necessary as reagents in this reaction include triphenylphosphine, tri-n-butylphosphine, tri-butylphosphine, tristolylphosphine, diphenyl-2-pyridylphosphine, and the like. Of these, triphenylphosphine and diphenyl-1-pyridylphosphine are preferred, and triphenylphosphine is more preferred.
[0042] ァゾ試薬の例としては、ジェチルァゾジカルボキシレート、ジイソプロピルァゾジカル ボキシレートゃジー tert—ブチルァゾジカルボキシレート、 1,1'ーァゾビス (N, N—ジ メチルホルムアミド)や 1, 1 ' - (ァゾジカルボ-ル)ジピペリジンを用いることができる。
中でも、ジェチルァゾジカルボキシレート(DEAD)、ジイソプロピルァゾジカルボキシ レートが好ましい。 [0042] Examples of azo reagents include jetyl azodicarboxylate, diisopropyl azodicarboxyloxy tert-butyl azodicarboxylate, 1,1'-azobis (N, N-dimethylformamide), 1,1 ′-(azodicarbol) dipiperidine can be used. Of these, jetylazodicarboxylate (DEAD) and diisopropylazodicarboxylate are preferable.
[0043] 本反応に用いる溶媒はテトラヒドロフラン、ジォキサン、トルエン、塩化メチレン、クロ 口ホルム、ァセトニトリル、酢酸ェチル、ジメチルスルホキシド、 N, N ジメチルホルム アミド等であり、好ましいのは、テトラヒドロフラン、トルエンである。 [0043] Solvents used in this reaction are tetrahydrofuran, dioxane, toluene, methylene chloride, chloroform, acetonitrile, ethyl acetate, dimethyl sulfoxide, N, N dimethylformamide, etc., preferably tetrahydrofuran and toluene.
[0044] 反応温度は 20°Cから室温が好ましぐ 5°Cから + 5°Cがより好ましい。 [0044] The reaction temperature is preferably from 20 ° C to room temperature, more preferably from 5 ° C to + 5 ° C.
(2)工程 2 (脱べンジル化) (2) Step 2 (Debenzylation)
上記で得られた化合物 3Aをパラジウム活性炭、水酸化パラジウム、又は白金ーパ ラジウム活性炭等の触媒を用いて水素雰囲気下にて接触水素添加することによりべ ンジル基を除去することができる。中でも水酸化パラジウムが触媒として好ましい。こ の反応に使用する溶媒としては、メタノール、エタノール、イソプロパノール、酢酸ェ チル、酢酸等を挙げることができる。反応温度は室温から還流温度であるが、室温が 好ましい。 The benzyl group can be removed by catalytic hydrogenation of the compound 3A obtained above using a catalyst such as palladium activated carbon, palladium hydroxide, or platinum-palladium activated carbon in a hydrogen atmosphere. Of these, palladium hydroxide is preferred as the catalyst. Examples of the solvent used in this reaction include methanol, ethanol, isopropanol, ethyl acetate, acetic acid and the like. The reaction temperature is from room temperature to reflux temperature, with room temperature being preferred.
(3)工程 3 (ァミノ基への変換) (3) Step 3 (Conversion to amino group)
上記で得られた化合物 4Aの水酸基をァミノ基に変換しィ匕合物 5Aを得ることができ る。まず、化合物 4Aをトリエチルァミン、 N ェチル N, N—ジイソプロピルァミン、 ピリジン、炭酸カリウム、炭酸カルシウム、炭酸セシウム等の塩基の存在下、メタンス ルホユルク口リドゃ p—トルエンスルホユルク口リドを用いて、脱離基を導入する。この 反応に使用する溶媒としては、クロ口ホルム、ジクロロメタン、ジェチルエーテル、テト ラヒドロフラン、ジメトキシェタン、酢酸ェチル等が挙げられる。反応温度は 0°Cから室 温である。 The compound 5A can be obtained by converting the hydroxyl group of the compound 4A obtained above into an amino group. First, compound 4A was used in the presence of a base such as triethylamine, Nethyl N, N-diisopropylamine, pyridine, potassium carbonate, calcium carbonate, cesium carbonate, etc. Then, a leaving group is introduced. Examples of the solvent used for this reaction include chloroform, dichloromethane, jetyl ether, tetrahydrofuran, dimethoxyethane, and ethyl acetate. The reaction temperature is from 0 ° C to room temperature.
[0045] 次に、アジィ匕ナトリウムを用いて、アジド基に変換することができる。この反応に使用 する溶媒としては、 N, N ジメチルホルムアミド、ジメチルスルホキシド、 N—メチルビ 口リドン、ジクロロメタン、ジェチルエーテル、テトラヒドロフラン、ジメトキシェタン、酢酸 ェチル等が挙げられる。反応温度は室温力 還流温度である。 [0045] Next, it can be converted to an azide group using azidosodium. Examples of the solvent used in this reaction include N, N dimethylformamide, dimethyl sulfoxide, N-methylbicycloidone, dichloromethane, jetyl ether, tetrahydrofuran, dimethoxyethane, and ethyl acetate. The reaction temperature is room temperature and reflux temperature.
[0046] さらに、アジド基を上記の脱べンジルイ匕の条件と同様な接触水素添カ卩にてアミノ基 に変換しィ匕合物 5Aが得られる。 [0046] Further, the azide group is converted to an amino group by catalytic hydrogenation similar to the above-mentioned debenzylation conditions to obtain compound 5A.
(4)工程 4
 P2、 P3及び P4の脱保護)
5—チォグルコースの保護基を適当な塩基を用いて除去し、本発明化合物 (I)が得 られる。この反応に使用する塩基として、ナトリウムメトキシド、ナトリウムエトキシド、ナ トリウムベンジルォキシド、水酸化ナトリウム、水酸化リチウム、炭酸カリウム、炭酸セシ ゥム、トリェチルァミン等が挙げられる。反応に適当な溶媒はメタノール、エタノール、 ベンジルアルコール、水又はこれらの混合溶媒である。反応温度は 0°C力 室温であ り、室温が好ましい。 (4) Process 4 Deprotection of P 2, P 3 and P 4) The protecting group for 5-thioglucose is removed using a suitable base to obtain the compound (I) of the present invention. Examples of the base used in this reaction include sodium methoxide, sodium ethoxide, sodium benzyloxide, sodium hydroxide, lithium hydroxide, potassium carbonate, cesium carbonate, triethylamine and the like. Suitable solvents for the reaction are methanol, ethanol, benzyl alcohol, water or a mixed solvent thereof. The reaction temperature is 0 ° C force room temperature, preferably room temperature.
P2、 P3及び P4の脱保護)
5—チォグルコースの保護基を適当な塩基を用いて除去し、本発明化合物 (I)が得 られる。この反応に使用する塩基として、ナトリウムメトキシド、ナトリウムエトキシド、ナ トリウムベンジルォキシド、水酸化ナトリウム、水酸化リチウム、炭酸カリウム、炭酸セシ ゥム、トリェチルァミン等が挙げられる。反応に適当な溶媒はメタノール、エタノール、 ベンジルアルコール、水又はこれらの混合溶媒である。反応温度は 0°C力 室温であ り、室温が好ましい。 (4) Process 4 Deprotection of P 2, P 3 and P 4) The protecting group for 5-thioglucose is removed using a suitable base to obtain the compound (I) of the present invention. Examples of the base used in this reaction include sodium methoxide, sodium ethoxide, sodium benzyloxide, sodium hydroxide, lithium hydroxide, potassium carbonate, cesium carbonate, triethylamine and the like. Suitable solvents for the reaction are methanol, ethanol, benzyl alcohol, water or a mixed solvent thereof. The reaction temperature is 0 ° C force room temperature, preferably room temperature.
[0047] 製造法 2 [0047] Production method 2
製造法 1の化合物 5Aにおいて、 Yが単結合である化合物 5Bは、より好ましくは、以 下の方法で合成できる。ただし、記号は前記と同義である。 In compound 5A of production method 1, compound 5B in which Y is a single bond is more preferably synthesized by the following method. However, the symbols are as defined above.
[0048] [化 3] [0048] [Chemical 3]

[0049] ここに、ピラゾール誘導体 1Bは国際公開 WO2004Z019958号に準拠して合成 することができる。 [0049] Here, pyrazole derivative 1B can be synthesized according to International Publication WO2004Z019958.
[0050] まず、製造法 1の工程 1に示した光延反応により、ピラゾール誘導体 1Bから化合物 3Bを合成することができる。 [0050] First, compound 3B can be synthesized from pyrazole derivative 1B by the Mitsunobu reaction shown in step 1 of production method 1.
(5)工程 5 (ニトロ基の還元) (5) Step 5 (Reduction of nitro group)
化合物 3Bをパラジウム活性炭、水酸化パラジウム、又は酸化白金等の触媒を用い て水素雰囲気下にて接触水素添加することにより-トロ基を還元することができる。中 でも酸化白金等の白金系触媒が好ましい。この反応に使用する溶媒としては、メタノ ール、エタノール、イソプロパノール、酢酸ェチル、酢酸等を挙げることができる。反
応温度は室温から還流温度であるが、室温が好ま U、。 The -tro group can be reduced by catalytic hydrogenation of Compound 3B under a hydrogen atmosphere using a catalyst such as palladium activated carbon, palladium hydroxide, or platinum oxide. Of these, platinum-based catalysts such as platinum oxide are preferred. Examples of the solvent used in this reaction include methanol, ethanol, isopropanol, ethyl acetate, and acetic acid. Anti The reaction temperature is from room temperature to reflux temperature, but room temperature is preferred.
[0051] 製造法 3 [0051] Production method 3
本発明の式 (I)化合物において、 Qが— N(RA)RBであって、 RAは- REであり、 RBは水素原子である化合物は以下の方法で合成できる。 In the compound of formula (I) of the present invention, a compound in which Q is —N (R A ) R B , R A is —R E , and R B is a hydrogen atom can be synthesized by the following method.
[0052] ただし、記号は前記と同義である。 [0052] However, the symbols are as defined above.
[0053] [化 4] [0053] [Chemical 4]

[0054] (6)工程 6 (グァニジノ基の導入) [0054] (6) Step 6 (Introduction of guanidino group)
化合物 5Aからグァ-ジノ化試薬 6Aを用いて化合物 7Aに誘導することができる。こ の反応に使用する溶媒としては、テトラヒドロフラン、 N, N—ジメチルホルムアミド、メ タノール、エタノール、イソプロパノール、酢酸ェチル、トルエン等が挙げられる。反応 温度は室温から還流温度である。 Compound 5A can be derivatized to compound 7A using guadinating reagent 6A. Examples of the solvent used in this reaction include tetrahydrofuran, N, N-dimethylformamide, methanol, ethanol, isopropanol, ethyl acetate, and toluene. The reaction temperature is from room temperature to reflux temperature.
(7)工程 7及び工程 8 (脱保護) (7) Step 7 and Step 8 (Deprotection)
グァ -ジノ基の保護基 RE、ベンジルォキシカルボ-ル基をナトリウムベンジルォキシ ドで処理することで、本発明化合物(I) (Q= - C ( = NH) NHCOOBn)が得られる。 さら〖こ、ベンジルォキシカルボニル基は、製造法 1の工程 2に記載した脱べンジルイ匕 と同様な方法で除去することができる。このときの好ましい触媒は水酸化パラジウムで める。 The compound (I) (Q = -C (= NH) NHCOOBn) of the present invention can be obtained by treating the protecting group R E of the gua-dino group and the benzyloxycarbonyl group with sodium benzyloxide. Furthermore, the benzyloxycarbonyl group can be removed by a method similar to the debenzylation method described in Step 2 of Production Method 1. The preferred catalyst at this time is palladium hydroxide.
製造法 4 Manufacturing method 4
本発明の式 (I)化合物において、 Qが— N(RA)RBであって、 RAは— CONHRD、 RB
は水素原子である(式中、 RDは水素原子または、水酸基及びピリジル基からなる群か ら選択される置換基で置換されてもょ 、C アルキル基を示す)化合物は以下の方 In the compound of formula (I) of the present invention, Q is —N (R A ) R B , and R A is —CONHR D , R B Is a hydrogen atom (wherein R D represents a C alkyl group which may be substituted with a hydrogen atom or a substituent selected from the group consisting of a hydroxyl group and a pyridyl group).
1-6 1-6
法で合成できる。ただし、下記スキーム中、他の記号は前記と同義である。 Can be synthesized. However, in the following schemes, other symbols are as defined above.
[0055] [化 5] [0055] [Chemical 5]

[0056] (8)工程 9 (ウレイド基の構築) [0056] (8) Step 9 (Construction of ureido group)
化合物 5Aから適当な塩基の存在下、 p 二トロフエ-ルクロロホルメート、トリホスゲ ンあるいは N, N' —カルボ-ルジイミダゾール(CDI)を用いて、ァミン RDNHと縮 Compound 5A is condensed with ammine R D NH using p-nitrophenyl chloroformate, triphosgene or N, N'-carbodiimidazole (CDI) in the presence of a suitable base.
2 合することにより、化合物 8Aに誘導することができる。適当な塩基としては、トリェチ ルァミン、 N ェチル N, N ジイソプロピルァミン、ピリジン、 DBU、炭酸カリウム、 炭酸カルシウム、炭酸セシウム等が挙げられる。また、使用する溶媒としては、クロ口 ホルム、ジクロロメタン、ジェチルエーテル、テトラヒドロフラン、 N, N ジメチルホル ムアミド、ァセトニトリル、酢酸ェチル等が挙げられる。反応温度は 0°Cから還流温度 である。 By combining these compounds, compound 8A can be derived. Suitable bases include triethylamine, Nethyl N, N diisopropylamine, pyridine, DBU, potassium carbonate, calcium carbonate, cesium carbonate and the like. Examples of the solvent used include chloroform, dichloromethane, jetyl ether, tetrahydrofuran, N, N dimethylformamide, acetonitrile, and ethyl acetate. The reaction temperature is from 0 ° C to the reflux temperature.
(9)工程 10 (脱保護) (9) Step 10 (Deprotection)
P2、 P3、及び P4を製造法 1の工程 4に記載の方法で脱保護し、本発明化合物 (I )が得られる。 P 2 , P 3 , and P 4 are deprotected by the method described in Step 4 of Production Method 1 to give compound (I) of the present invention.
[0057] 製造法 5 [0057] Manufacturing method 5
本発明の式 (I)化合物において、 Qがー N(RA)RBであって、 RA及び RBは同一また は異なって、水素原子、または、 C アルキル基 (該 C アルキル基は、水酸基、 C
ONH及び OC アルキルフ -ルカ なる群力 選択される置換基で置換されてIn the compound of the formula (I) of the present invention, Q is —N (R A ) R B , and R A and R B are the same or different and are a hydrogen atom or a C alkyl group (the C alkyl group is , Hydroxyl group, C ONH and OC alkyl-Fluca are group forces that are substituted with selected substituents
2 1-4 2 1-4
もよい)である化合物は以下の方法で合成できる。下記スキーム中、 L1はハロゲン原 子、 MeSO 0-等の脱離基を示し、その他の記号は前記と同義である。 May be synthesized by the following method. In the following scheme, L1 represents a halogen atom, a leaving group such as MeSO 0-, and the other symbols are as defined above.
[0058] [ィ匕 6] [0058] [6]

[0059] (10)工程 11 [0059] (10) Step 11
化合物 5Aを適当な塩基の存在下、 RAL1又は RBL1と反応させることにより、化合 物 9Aが得られる。適当な塩基としては、トリェチルァミン、 N ェチルー N, N ジィ ソプロピルァミン、ピリジン、 DBU、炭酸カリウム、炭酸カルシウム、炭酸セシウム等が 挙げられる。また、使用する溶媒としては、 N, N ジメチルホルムアミド、ァセトニトリ ル、メタノール、エタノール、クロロホノレム、ジクロロメタン、ジェチノレエーテノレ、テトラヒ ドロフラン等が挙げられる。反応温度は室温力 還流温度である。 Compound 9A is obtained by reacting compound 5A with R A L1 or R B L1 in the presence of a suitable base. Suitable bases include triethylamine, N ethyl N, N disopropylamine, pyridine, DBU, potassium carbonate, calcium carbonate, cesium carbonate and the like. Examples of the solvent to be used include N, N dimethylformamide, acetonitrile, methanol, ethanol, chlorophenol, dichloromethane, jetinoreethenole, tetrahydrofuran and the like. The reaction temperature is room temperature and reflux temperature.
(11)工程 12 (11) Process 12
化合物 4Aから製造法 1のァミノ基への変換と同様の条件で、化合物 9Aを製造する こともできる。まず、化合物 4Aをトリエチルァミン、 N ェチル N, N ジイソプロピ ルァミン、ピリジン、炭酸カリウム、炭酸カルシウム、炭酸セシウム等の塩基の存在下、 メタンスルホユルク口リドゃ p—トルエンスルホユルク口リドを用いて、脱離基を導入す る。この反応に使用する溶媒としては、クロ口ホルム、ジクロロメタン、ジェチルエーテ ル、テトラヒドロフラン、ジメトキシェタン、酢酸ェチル等が挙げられる。反応温度は 0 °Cから室温である。 Compound 9A can also be produced under the same conditions as for conversion from compound 4A to the amino group of production method 1. First, compound 4A was prepared using methanesulfuryl chloride in the presence of a base such as triethylamine, Nethyl N, N diisopropylamine, pyridine, potassium carbonate, calcium carbonate, cesium carbonate, etc. using p-toluenesulfuryl chloride. Introduce a leaving group. Examples of the solvent used for this reaction include black mouth form, dichloromethane, jetyl ether, tetrahydrofuran, dimethoxyethane, and ethyl acetate. The reaction temperature is from 0 ° C to room temperature.
[0060] 次に、トリエチノレアミン、 N ェチノレ一 N, N ジイソプロピルァミン、ピリジン、 DBU
、炭酸カリウム、炭酸カルシウム、炭酸セシウム等の適当な塩基の存在下、 NHRARB と反応させることによりィ匕合物 9Aが得られる。この反応に使用する溶媒としては、 N, N—ジメチルホルムアミド、ジメチルスルホキシド、 N—メチルピロリドン、ジクロロメタン 、ジェチルエーテル、テトラヒドロフラン、ジメトキシェタン、酢酸ェチル等が挙げられ る。反応温度は室温から還流温度である。 [0060] Next, triethinoreamine, N ethinorei N, N diisopropylamine, pyridine, DBU Compound 9A is obtained by reacting with NHR A R B in the presence of a suitable base such as potassium carbonate, calcium carbonate, cesium carbonate or the like. Examples of the solvent used in this reaction include N, N-dimethylformamide, dimethyl sulfoxide, N-methylpyrrolidone, dichloromethane, jetyl ether, tetrahydrofuran, dimethoxyethane, and ethyl acetate. The reaction temperature is from room temperature to reflux temperature.
(12)工程 13 (脱保護) (12) Step 13 (Deprotection)
P2、 P3及び P4を製造法 1の工程 4に記載の方法で脱保護し、本発明化合物 (I) が得られる。 P 2 , P 3 and P 4 are deprotected by the method described in Step 4 of Production Method 1 to give the compound (I) of the present invention.
製造法 6 Manufacturing method 6
本発明の式 (I)化合物において、 Qが— CONHReであり、 Yが C アルキレン基、 In the compound of formula (I) of the present invention, Q is —CONHR e , Y is a C alkylene group,
2-6 2-6
又は C ァルケ-レン基である化合物は以下の方法で合成できる。 Alternatively, a compound having a C alkellene group can be synthesized by the following method.
2-6 2-6
[0061] ただし、 Y1は単結合、又は C アルキレン基を示し、その他の記号は前記と同義で [0061] However, Y 1 represents a single bond or a C alkylene group, and other symbols are as defined above.
1-4 1-4
ある。 is there.
[0062] [化 7] [0062] [Chemical 7]



[0064] まず、製造法 1の工程 1に示した光延反応により、ピラゾール誘導体 1Cから化合物 3Cを合成することができる。 First, the compound 3C can be synthesized from the pyrazole derivative 1C by the Mitsunobu reaction shown in Step 1 of Production Method 1.
( 13)工程 14 (Heck反応) (13) Process 14 (Heck reaction)
化合物 3Cとォレフイン酢酸 10Aをパラジウム触媒とホスフィンリガンド、及び適当な 塩基の存在下、 Heck反応を行うことによりィ匕合物 11Aを合成することができる。このと き用いるパラジウム触媒としては、酢酸パラジウム、テトラキストリフエ-ルホスフィンパ ラジウム、ジベンジリデンアセトンパラジウム、ビストリフエ-ルホスフィンパラジウムクロ リド、パラジウム活性炭等が挙げられる。ホスフィンリガンドとしてはトリフエニルホスフィ ンゃトリス (2—メチルフエ-ル)ホスフィン等が挙げられる。また、塩基にはトリエチルァ ミン、 N—ェチルー N, N—ジイソプロピルァミン、炭酸カリウム、炭酸カルシウム、炭 酸セシウム、カリウム t—ブトキシド等が用いられる。反応に用いられる溶媒としては、 ァセトニトリル、トルエン、テトラヒドロフラン等が挙げられる。反応温度は 0°Cから還流 温度であるが、マイクロウエーブを用いることもある。 Compound 11A can be synthesized by subjecting compound 3C and olefin acetic acid 10A to a Heck reaction in the presence of a palladium catalyst, a phosphine ligand, and an appropriate base. Examples of the palladium catalyst used at this time include palladium acetate, tetrakistriphenylphosphine palladium, dibenzylideneacetone palladium, bistriphenylphosphine palladium chloride, palladium activated carbon and the like. Examples of the phosphine ligand include triphenylphosphine tris (2-methylphenol) phosphine. As the base, triethylamine, N-ethyl-N, N-diisopropylamine, potassium carbonate, calcium carbonate, cesium carbonate, potassium t-butoxide and the like are used. Examples of the solvent used in the reaction include acetonitrile, toluene, tetrahydrofuran and the like. The reaction temperature ranges from 0 ° C to the reflux temperature, but a microwave may be used.
( 14)工程 15 (アミド化) (14) Step 15 (Amidation)
化合物 11八とァミン( ?《1 )にて脱水縮合し、化合物 12Aが得られる。この反応 Compound 12A is obtained by dehydration-condensation with Compound 11-8 and amine (? << 1). This reaction
2 2
に使用する溶媒としては、クロ口ホルム、ジクロロメタン、 N, N—ジメチルホルムアミド 等が好ましぐ脱水縮合剤としては、 N, N '—ジシクロへキシルカルポジイミド (DCC) 、 N—ェチル— N,— 3—ジメチルァミノプロピルカルボジイミド塩酸 (WSC)、 CDI、 WSCZl—ヒドロキシベンゾトリアゾール 1水和物等が好まし 、。ここでの反応温度は 0°C〜60°Cである。 As the solvent used in the reaction, chloroform, formaldehyde, dichloromethane, N, N-dimethylformamide and the like are preferred. As the dehydrating condensing agent, N, N′-dicyclohexylcarpositimide (DCC), N-ethyl-N, — 3-Dimethylaminopropylcarbodiimide hydrochloride (WSC), CDI, WSCZl—hydroxybenzotriazole monohydrate, etc. are preferred. The reaction temperature here is 0 ° C to 60 ° C.
( 15)工程 16 (15) Process 16
化合物 11Aのォレフイン部分を製造法 1の工程 2に示した接触水素添加を行った 後に、ァミン (ReNH )と縮合することで、化合物 12Bを製造することもできる。 Compound 12B can also be produced by condensing the olefin moiety of Compound 11A with catalytic amine addition shown in Step 2 of Production Method 1 and then condensing with ammine (R e NH 3).
2 2
( 16)工程 17 (脱保護) (16) Step 17 (Deprotection)
最後に、
 P2、 P3及び P4を製造法 1の工程 4に示した方法で脱保護し、本発明化 合物 (I)が得られる。
[0065] 製造法 7 Finally, P 2 , P 3 and P 4 are deprotected by the method shown in Step 4 of Production Method 1 to obtain the compound (I) of the present invention. [0065] Production method 7
P2、 P3及び P4を製造法 1の工程 4に示した方法で脱保護し、本発明化 合物 (I)が得られる。
[0065] 製造法 7 Finally, P 2 , P 3 and P 4 are deprotected by the method shown in Step 4 of Production Method 1 to obtain the compound (I) of the present invention. [0065] Production method 7
本発明の式 (I)化合物において、 Qが— CONHReであり、 Yが— O— (CH ) n—で In the compound of formula (I) of the present invention, Q is —CONHR e , Y is —O— (CH 2) n—
2 ある化合物は以下の方法で合成できる。 2 A compound can be synthesized by the following method.
[0066] ただし、 Y1は C アルキレン基を示し、 L2はハロゲン原子、 MeSO O-等の脱離基 [0066] However, Y 1 represents a C alkylene group, L2 represents a leaving group such as a halogen atom, MeSO 2 O-, etc.
1-4 2 1-4 2
を示し、その他の記号は前記と同義である。 The other symbols are as defined above.
[0067] [化 8] [0067] [Chemical 8]

[0068] ここに、ピラゾール誘導体 4Bは製造法 1の工程 1、工程 2に準拠して合成することが できる。 [0068] Here, pyrazole derivative 4B can be synthesized in accordance with steps 1 and 2 of production method 1.
( 17)工程 18 (17) Process 18
ピラゾール誘導体 4Bを適当な塩基の存在下で、化合物 13 Aと反応させることにより 化合物 12Cを得ることができる。適当な塩基としては水素化ナトリウム、炭酸カリウム、 炭酸セシウム、炭酸ナトリウム、 n—ブチルリチウムが好ましぐこの反応に使用する溶 媒としては、テトラヒドロフラン、ジェチルエーテル、 N, N—ジメチルホルムアミド、ァ セトン、 DMSOが好ましい。ここでこの反応の温度は 0°C〜60°Cである。 Compound 12C can be obtained by reacting pyrazole derivative 4B with compound 13A in the presence of a suitable base. Sodium hydride, potassium carbonate, cesium carbonate, sodium carbonate, and n-butyllithium are preferred as suitable bases. Solvents used in this reaction include tetrahydrofuran, jetyl ether, N, N-dimethylformamide, Seton and DMSO are preferred. Here, the temperature of this reaction is 0 ° C to 60 ° C.
( 18)工程 19 (18) Process 19
P2、 P3及び P4を製造法 1の工程 4に示した方法で脱保護し、本発明化合物 (I) が得られる。
[0069] 本発明の化合物は、 SGLT1を選択的に阻害し、小腸からの糖の吸収を抑制して、P 2 , P 3 and P 4 are deprotected by the method shown in Step 4 of Production Method 1 to obtain the compound (I) of the present invention. [0069] The compound of the present invention selectively inhibits SGLT1, suppresses absorption of sugar from the small intestine,
IGTを改善することができる。 IGT can be improved.
[0070] よって、本発明の化合物は、 SGLT1阻害剤、あるいは、糖尿病、糖尿病関連疾患 及び糖尿病合併症の予防又は治療剤の有効成分として用いることができる。 Therefore, the compound of the present invention can be used as an active ingredient of an SGLT1 inhibitor or a preventive or therapeutic agent for diabetes, diabetes-related diseases and diabetic complications.
[0071] ここで、「糖尿病」には、 1型糖尿病、 2型糖尿病の他、特定の原因によるその他の 型の糖尿病が含まれる。 Here, “diabetes” includes type 1 diabetes, type 2 diabetes, and other types of diabetes caused by a specific cause.
[0072] ここで、「糖尿病関連疾患」とは、肥満、高インスリン血症、糖代謝異常、高脂質血 症、高コレステロール血症、高トリグリセリド血症、脂質代謝異常、高血圧、うつ血性心 不全、浮腫、高尿酸血症、痛風などが挙げられる。 [0072] Here, "diabetes-related disease" refers to obesity, hyperinsulinemia, abnormal glucose metabolism, hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, abnormal lipid metabolism, hypertension, congestive heart failure Edema, hyperuricemia, gout and the like.
[0073] ここで、「糖尿病合併症」は、急性合併症及び慢性合併症に分類される。 Here, “diabetic complications” are classified into acute complications and chronic complications.
[0074] 「急性合併症」には、高血糖 (ケトアシドーシスなど)、感染症 (皮膚、軟部組織、胆 道系、呼吸系、尿路感染など)などが挙げられる。 [0074] "Acute complications" include hyperglycemia (such as ketoacidosis), infection (such as skin, soft tissue, biliary system, respiratory system, urinary tract infection) and the like.
[0075] 「慢性合併症」には、細小血管症(腎症、網膜症)、動脈硬化症 (ァテローム性動脈 硬化症、心筋梗塞、脳梗塞、下肢動脈閉塞など)、神経障害 (感覚神経、運動神経、 自律神経など)、足壊疽などが挙げられる。 [0075] “Chronic complications” include microangiopathy (nephropathy, retinopathy), arteriosclerosis (eg, atherosclerosis, myocardial infarction, cerebral infarction, lower limb arterial occlusion), neuropathy (sensory nerve, Motor nerves, autonomic nerves, etc.) and foot gangrene.
[0076] 主要な合併症は、糖尿病網膜症、糖尿病腎症、糖尿病神経障害である。 [0076] The major complications are diabetic retinopathy, diabetic nephropathy, diabetic neuropathy.
[0077] 本発明の化合物は、医薬として、全身的又は局所的に、経口投与又は非経口投与 することができる。 [0077] The compound of the present invention can be administered as a medicine systemically or locally, orally or parenterally.
[0078] 本発明化合物は、該化合物の作用の増強または該化合物の投与量の低減などを 目的として、 SGLT1及び SGLT2活性阻害薬以外のことなった作用機序の糖尿病 治療剤、糖尿病性合併症治療剤、抗高脂血症剤、降圧剤、抗肥満剤、利尿剤、抗 血栓剤などの薬剤(以下、併用薬剤と略記する)と組み合わせて用いることができる 。この際、本発明化合物と併用薬剤の投与時期は限定されず、これらを投与対象に 対し、同時に投与してもよいし、時間差をおいて投与してもよい。さらに、本発明化合 物と併用薬剤とは、それぞれの活性成分を含む 2種類の製剤として投与されてもょ ヽ し、両方の活性成分を含む単一の製剤として投与されてもよい。併用薬剤の投与量 は、臨床上用いられている用量を基準として適宜選択することができる。また、本発 明化合物と併用薬剤の配合比は、投与対象、投与ルート、対象疾患、症状、組み合
わせなどにより適宜選択することができる。例えば、投与対象がヒトである場合、本発 明化合物 1重量部に対し、併用薬剤を 0. 01〜: LOO重量部用いればよい。 [0078] The compound of the present invention is a therapeutic agent for diabetes and a diabetic complication having a different mechanism of action other than SGLT1 and SGLT2 activity inhibitors for the purpose of enhancing the action of the compound or reducing the dose of the compound. It can be used in combination with drugs (hereinafter abbreviated as concomitant drugs) such as therapeutic agents, antihyperlipidemic agents, antihypertensive agents, antiobesity agents, diuretics and antithrombotic agents. At this time, the administration time of the compound of the present invention and the concomitant drug is not limited, and these may be administered simultaneously to the administration subject or may be administered with a time difference. Furthermore, the compound of the present invention and the concomitant drug may be administered as two types of preparations containing each active ingredient, or may be administered as a single preparation containing both active ingredients. The dose of the concomitant drug can be appropriately selected based on the clinically used dose. The compounding ratio of the compound of the present invention and the concomitant drug is the subject of administration, administration route, target disease, symptoms, combination It can be appropriately selected depending on the combination. For example, when the administration subject is a human, the concomitant drug may be used in an amount of 0.01 to LOO parts by weight per 1 part by weight of the compound of the present invention.
なお、糖尿病治療剤としては、例えばインスリン製剤(例、ゥシ、ブタの脾臓力ゝら抽 出された動物インスリン製剤;大腸菌またはイーストを用い、遺伝子工学的に合成し たヒトインスリン製剤;インスリン亜 プロタミンインスリン亜 インスリンのフラグメン トまたは誘導体 (例、 INS— 1等)、経口インスリン製剤)、インスリン抵抗性改善剤 (例 Examples of diabetes therapeutic agents include insulin preparations (eg, animal insulin preparations extracted from sushi and swine spleen; human insulin preparations synthesized using E. coli or yeast and genetically engineered; Protamine insulin sub-insulin fragments or derivatives (eg, INS-1 etc.), oral insulin preparations), insulin resistance improvers (eg,
、ピオグリタゾンまたはその塩 (好ましくは塩酸塩)、ロシグリタゾンまたはその塩 (好ま しくはマレイン酸塩) 、リボグリタゾン (Rivoglitazone) (CS— Oi l) (R— 119702)、 シポグリタザール(Sipoglitazar) (TAK—654)、メタグリダセン (Metaglidasen) (M XB- 1 0 2)、ナベグリタザール (Naveglitazar) (LY— 519818)、 MX— 6054、ノ ラグリタゾン (Balaglitazone) (NN— 2344)、 T— 131 (AMG131)、 PPAR y ァゴ -スト、 PPAR yアンタゴ-スト、 PPAR y / aデュアルァゴ-スト、 a—ダルコシダー ゼ阻害剤(例、ボグリボース、ァカルボース、ミグリトール、エミダリテート)、ビグアナィ ド剤(例、フェンホルミン、メトホルミン、ブホルミンまたはそれらの塩 (例、塩酸塩、フマ ル酸塩、コハク酸塩))、インスリン分泌促進剤 (スルホ-ルゥレア剤(例、トルプタミド、 ダリベンクラミド、ダリクラジド、クロルプロパミド、トラザミド、ァセトへキサミド、ダリクロピ ラミド、グリメピリド、ダリピザイド、ダリブゾール等)、レパグリニド、セナグリニド、ナテグ リニド、ミチグリニドまたはそのカルシウム塩水和物) 、 GPR40ァゴ-スト、 GPR40ァ ンタゴ-スト、 GLP— 1受容体ァゴ-スト(例、 GLP— 1、 GLP— 1MR剤、リラグルチ ド(Liraglutide) (NN— 2211)、 Exenatide(AC - 2993) (exendin— 4)、 Exenati de LAR、: BIM— 51077、 A i b (8, 35) hGLP— 1(7, 37) NH2、 CJC— 1131、 A VE0010、 GSK— 716155)、アミリンァゴニスト(例、プラムリンチド) 、フォスフォチ口 シンフォスファターゼ阻害剤(例、バナジン酸ナトリウム) 、ジぺプチジルぺプチダー ゼ I V阻害剤(例、 WO02/038541に記載のィ匕合物、 NVP— DPP— 278、 PT— 100、 P32/98,ビルダグジプチン (Vildagliptin) (LAF— 237)、 P93Z01、シタグ リプチン (SitagliptinXMK— 431)、サクサグリプチン(Saxagliptin) (BMS— 4771 18)、 SYR— 322、 MP— 513、 T— 6666、 GRC— 8200等)、 j8 3ァゴ-スト(例、 A J 9677、 AZ40140等) 、糖新生阻害剤(例、グリコーゲンホスホリラーゼ阻害剤、
グルコース一 6 ホスファターゼ阻害剤、グルカゴン拮抗剤、フルクトース一 1, 6 ビ スホスファターゼ阻害剤)、 SGLT (sodium-glucose cotransporter) 阻害剤( ί列、 WO04/014931, WO04/089967, WO06/073197に記載のィ匕合物、 Τ 1 095、 Sergliflozin (GSK- 869682)、 GSK— 189075、 KGT— 1251、 KGT— 1 681、 KGA— 2727、 BMS— 512148、 AVE2268、 SAR7226等)、 1 I j8—ヒドロ キシステロイドデヒドロゲナーゼ阻害薬(例、 WO06/51662に記載のィ匕合物、 BVT — 3498、 INCB13739)、 GPR119ァゴ-スト(例、 PSN— 632408、 APD— 668) 、アディポネクチンまたはその作動薬、 IKK阻害薬(例、 A S— 2868)、 AMPK活性 化薬、レブチン抵抗性改善薬、ソマトスタチン受容体作動薬、ダルコキナーゼ活性化 薬(例、 Ro— 28— 1675)、脾リパーゼ阻害薬(例、オルリスタツト、 ATL—962)、 D GAT— 1阻害薬が挙げられる。 Pioglitazone or its salt (preferably hydrochloride), rosiglitazone or its salt (preferably maleate), riboglitazone (CS—Oi l) (R—119702), Sipoglitazar (TAK— 654), Metaglidasen (M XB- 1 0 2), Naveglitazar (LY—519818), MX—6054, Nolaglitazone (NN—2344), T—131 (AMG131), PPAR y Ago-st, PPAR y-antagost, PPAR y / a dual-agost, a-Darcosidase inhibitor (eg, voglibose, carbolose, miglitol, emidalitate), biguanide (eg, phenformin, metformin, buformin or Their salts (eg, hydrochloride, fumarate, succinate)), insulin secretagogues (sulfururea (eg, tolptamide, daribenclamide, da Liclazide, chlorpropamide, tolazamide, acetohexamide, daliclopyramide, glimepiride, dalipizide, dalibuzole, etc.), repaglinide, senaglinide, nateglinide, mitiglinide or its calcium salt hydrate), GPR40 agonist, GPR40 antagonist , GLP-1 receptor agonist (eg, GLP-1, GLP-1 MR agent, Liraglutide (NN-2211), Exenatide (AC-2993) (exendin-4), Exenati de LAR, BIM— 51077, A ib (8, 35) hGLP— 1 (7, 37) NH2, CJC— 1311, A VE0010, GSK— 716155), amylin agonist (eg, pramlintide), phosphochiphosphatase Synphosphatase inhibitor (eg , Sodium vanadate), dipeptidyl peptidase IV inhibitor (eg, a compound described in WO02 / 038541, NVP—DPP—278, PT—100, P32 / 98, Vildagliptin (LAF — 237), P93Z01, sitagliptin (SitagliptinXMK—431), saxagliptin (BMS—4771 18), SYR—322, MP—513, T—6666, GRC—8200, etc.), j8 3agost ( Eg, AJ 9677, AZ40140, etc.), gluconeogenesis inhibitors (eg, glycogen phosphorylase inhibitors, Glucose-6 phosphatase inhibitor, glucagon antagonist, fructose-1,6 bisphosphatase inhibitor), SGLT (sodium-glucose cotransporter) inhibitor (described in ί row, WO04 / 014931, WO04 / 089967, WO06 / 073197) Compound, Τ 1 095, Sergliflozin (GSK-869682), GSK—189075, KGT—1251, KGT—1 681, KGA—2727, BMS—512148, AVE2268, SAR7226, etc.), 1 I j8—hydroxysteroid Dehydrogenase inhibitors (eg, compounds described in WO06 / 51662, BVT-3498, INCB13739), GPR119 agonist (eg, PSN-632408, APD-668), adiponectin or agonists thereof, IKK inhibitors (Eg, AS-2868), AMPK activator, levtin resistance improver, somatostatin receptor agonist, darcokinase activator (eg, Ro-28-1675), spleen lipase inhibitor (eg, orlistat, ATL) —962), D GAT-1 inhibitor.
[0080] 糖尿病性合併症治療剤としては、例えばアルドース還元酵素阻害剤(例、トルレス タツト、ェパノレレスタツト、ゼナレスタツト、ゾポノレレスタツト、ミナノレレスタツト、フィダレス タツト、 CT— 112)、神経栄養因子およびその増加薬(例、 NGF、 NT— 3、 BDNF、 ニューロトロフィン産生'分泌促進剤)、神経再生促進薬 (例、 Y—128)、 PKC阻害 剤(例、ルボキシスタウリンメシレート (ruboxistaurin mesylate; LY— 333531))、 AGE阻害剤(例、 ALT946、ピマゲジン、ピラトキサチン、 N—フエナシルチアゾリゥ ムブロマイド(ALT766)、 ALT— 711、 EXO— 226、ピリドリン(Pyridorin)、ピリド キサミン)、活性酸素消去薬 (例、チォタト酸)、脳血管拡張剤 (例、チアプリド、メキシ レチン)、ソマトスタチン受容体作動薬(例、 BIM23190)、アポトーシスシグナルレギ ユレ一ティングキナーゼ 1 (ASK— 1) 阻害薬が挙げられる。 [0080] As an agent for treating diabetic complications, for example, an aldose reductase inhibitor (eg, torres tart, epanolorestat, zenarestat, zoponolestat, minanorestat, fidarestat, CT-112) , Neurotrophic factor and its increasing drug (eg, NGF, NT-3, BDNF, neurotrophin production 'secretion promoter), nerve regeneration promoter (eg, Y-128), PKC inhibitor (eg, ruboxis) Taurine mesylate (LY- 333531)), AGE inhibitors (eg, ALT946, pimagedin, pyratoxatin, N-phenacyl thiazolium bromide (ALT766), ALT-711, EXO-226, pyridorin) , Pyridoxamine), active oxygen scavengers (eg, thiotate), cerebral vasodilators (eg, thiopride, mexiletine), somatostatin receptor agonists (eg, BIM23190), apoptotic sigma Ruregi Jure one computing kinase 1 (ASK 1) inhibitors.
[0081] 抗高脂血症剤としては、例えばスタチン系化合物(例、プラバスタチン、シンパスタ チン、口パスタチン、アトノレパスタチン、フルパスタチン、イタパスタチン、ロスバスタチ ン、ピタパスタチンまたはそれらの塩(例、ナトリウム塩、カルシウム塩)) 、スクアレン 合成酵素阻害剤(例、 TAK— 475)、フイブラート系化合物 (例、ベザフイブラート、 クロフイブラート、シムフイブラート、クリノフイブラート)、 AC AT阻害剤(例、アバシマ イブ(Avasimibe)、エフルシマイブ(Eflucimibe) )、陰イオン交換榭脂(例、コレス チラミン)、プロブコーノレ、ニコチン酸系薬剤(例、ニコモーノレ (nicomol)、ニセリトロー
ル (niceritrol))、ィコサペント酸ェチル、植物ステロール(例、ソイステロール (soyste rol)、ガンマオリザノール(γ—oryzanol))、 CETP阻害薬(例、 Torcetrapib、 JTT 705、JTT 302、 FN— VP4等)、コレステロール吸収抑制薬(例、ェゼチミブ(E zetimibe)等)が上げられる。 [0081] Antihyperlipidemic agents include, for example, statin compounds (eg, pravastatin, sympastatin, oral pastatin, atonolepastatin, flupastatin, itapastatin, rosbastatin, pitapastatin or salts thereof (eg, Sodium salts, calcium salts)), squalene synthase inhibitors (eg, TAK-475), fibrate compounds (eg, bezafibrate, clofibrate, simfibrate, clinofibrate), AC AT inhibitors (eg, abashimibe ( Avasimibe), Eflucimibe), anion exchange scab (eg, cholestyramine), probuconole, nicotinic acid drugs (eg, nicomol), niceritro Niceritrol), icosapentate ethyl, plant sterols (eg, soysterol, gamma-oryzanol), CETP inhibitors (eg, Torcetrapib, JTT 705, JTT 302, FN—VP4, etc.) And cholesterol absorption inhibitors (eg, ezetimibe).
[0082] 降圧剤としては、例えばアンジォテンシン変換酵素阻害剤(例、カプトプリル、ェナ ラプリル、デラプリル) 、アンジォテンシン II拮抗剤(例、カンデサルタン シレキセチ ル、口サルタン、ェプロサルタン、バルサルタン、テルミサルタン、ィルベサルタン、タ ソサルタン、アジルザルタン(TAK— 536) )、カルシウム拮抗剤(例、マ-ジピン、二 フエジピン、アムロジピン、エホニジピン、二カノレジピン)、カリウムチャンネノレ開口薬( 例、レブクロマカリム、 L 27152、 AL0671、 NIP— 121)、クロ-ジンが挙げられる [0082] Examples of the antihypertensive agent include angiotensin converting enzyme inhibitors (eg, captopril, enalapril, delapril), angiotensin II antagonists (eg, candesartan cilexetil, oral sultan, eprosartan, valsartan, telmisartan, Ilbesartan, tasosartan, azilsartan (TAK-536)), calcium antagonists (eg, mardipine, difedipine, amlodipine, efonidipine, dicanodipine), potassium channelnore openers (eg, lebucromacarim, L 27152, AL0671, NIP — 121), including clodin
[0083] 抗肥満剤としては、例えば中枢性抗肥満薬 (例、デキスフェンフルラミン、フェンフ ルラミン、フェンテルミン、シブトラミン、アンフエプラモン、デキサンフエタミン、マジン ドール、フエ-ルプロパノールァミン、クロべンゾレックス; MCH受容体拮抗薬(例、 W 006/035967に記載のィ匕合物、 SB— 568849 ; SNAP— 7941、 T 226296); ニューロペプチド 拮抗薬 (例、 CP— 422935);カンナピノイド受容体拮抗薬(例、 リモナノ ン卜(Rimonabant) (SR— 141716)、 SR— 147778) ;グレリン拮抗薬; 1 1 β—ヒドロキシステロイドデヒドロゲナーゼ阻害薬(例、 BVT— 3498、 INCB1373 9) )、脾リパーゼ阻害薬 (例、オルリスタツト、 ATL— 962)、 DGAT—1阻害薬、 /3 3 ァゴ-スト(例、 AJ— 9677、 AZ40140)、ペプチド性食欲抑制薬(例、レプチン、 CN TF (毛様体神経栄養因子))、コレシストキュンァゴ-スト(例、リンチトリブト、 FPL— 15849)、摂食抑制薬 (例、 P— 57)が挙げられる。 [0083] Anti-obesity agents include, for example, central anti-obesity agents (eg, dexfenfluramine, phenfluramine, phentermine, sibutramine, amphepramone, dexamphetamine, mazindol, phenolpropanolamine, clobenzolex). MCH receptor antagonists (eg, compounds described in W 006/035967, SB—568849; SNAP—7941, T 226296); Neuropeptide antagonists (eg, CP—422935); Cannapinoid receptor antagonists (Eg, Rimonabant (SR—141716), SR—147778); ghrelin antagonists; 1 1 β-hydroxysteroid dehydrogenase inhibitors (eg, BVT-3498, INCB1373 9)), spleen lipase inhibitors ( Eg, orlistat, ATL—962), DGAT—1 inhibitor, / 3 3 agonist (eg, AJ—9677, AZ40140), peptidic appetite suppressant (eg, leptin, CN TF (ciliary neurotrophic) factor)) , Cholecyst kyungagost (eg, Lynch tribute, FPL-15849), antifeedant (eg, P-57).
[0084] 利尿剤としては、例えば、キサンチン誘導体(例、サリチル酸ナトリウムテオプロミン 、サリチル酸カルシウムテオプロミン)、チアジド系製剤(例、ェチアジド、シクロペンチ アジド、トリクロルメチアジド、ヒドロクロ口チアジド、ヒドロフルメチアジド、ベンチルヒド 口クロ口チアジド、ペンフルチジド、ポリチアジド、メチクロチアジド)、抗アルドステロン 製剤 (例、スピロノラタトン、トリアムテレン)、炭酸脱水酵素阻害剤 (例、ァセタゾラミド) 、クロルベンゼンスルホンアミド系製剤(例、クロルタリドン、メフルシド、インダパミド)、
ァゾセミド、イソソルビド、エタクリン酸、ピレタ -ド、ブメタ-ド、フロセミドが挙げられる [0084] Examples of diuretics include xanthine derivatives (eg, sodium salicylate theopromine, calcium salicylate theopromine), thiazide preparations (eg, ethiazide, cyclopenthiazide, trichloromethiazide, hydrothiazide, hydroflumethiazide, benzilhydride. Mouth-mouth mouth thiazide, penflutide, polythiazide, methycrothiazide), anti-aldosterone preparation (eg, spironolatatone, triamterene), carbonic anhydrase inhibitor (eg, acetazolamide), chlorobenzenesulfonamide preparation (eg, chlorthalidone, mefluside, indapamide) , Examples include fazosemide, isosorbide, ethacrynic acid, pyrethradide, bumetide, and furosemide.
[0085] 抗血栓剤としては、例えば、へパリン (例、へパリンナトリウム、へパリンカルシウム、 ダルテパリンナトリウム (dalteparin sodium), AVE - 5026) 、ヮルフアリン(例、ヮ ルフアリンカリウムなど) 、抗トロンビン薬(例、アルガトロバン (argatroban)、キシメラ ガトフン (Ximelagatran入ダビ'ガトフン (Dabigatran入 Odiparcil、 Lepirudin、 bival irudin、 Desirudin、 ART— 123、 Idraparinux、 SR— 123781、 AZD— 0837、 M CC— 977、 TGN— 255、 TGN— 167、 RWJ— 58436、 LB— 30870、 MPC— 09 20、 Pegmusirudinゝ Org— 426751等)、血栓溶解薬(例、ゥロキナーゼ (urokina se)、チソキナーゼ (tisokinase)、ァノレテプラーゼ (alteplase)、ナテプラーゼ (natepla se)、モンテプラーゼ (monteplase)、パミテプラーゼ (pamiteplase)等)、血小板凝集 抑制薬(例、塩酸チクロピジン (ticlepidine hydrochloride),シロスタゾール(cilost azol)、ィコサペント酸ェチノレ、ベラプロストナトリウム (beraprost sodium),塩酸サノレ ポグレフート (sarpogrelate hydrochloride)など)、 Xa阻害薬 (例、 Fondaparinu x、 BAY— 59— 7939、 DU— 176b、 YM— 150、 SR— 126517、 Apixaban, Raz axaban, LY— 517717、 MLN— 102、 Octaparine、 Otamixaban, EMD— 503 982、 TC— 10、 CS— 3030、 AVE— 3247、 GSK— 813893、 KFA— 1982等)、 血漿中カルボキシぺプチターゼ B (または活性型 thrombin—activatable fibrinol ysis inhibitor [TAFIa]としても知られている)阻害薬(例、 AZD— 9684、 EF— 62 65、 MN— 462)などが挙げられる。 [0085] Antithrombotic agents include, for example, heparin (eg, heparin sodium, heparin calcium, dalteparin sodium, AVE-5026), sulfarin (eg, sulfarin potassium), antithrombin Drugs (eg, argatroban, xymeragatran (Dimigatran), Dabigatran (Odiparcil, Lepirudin, bival irudin, Desirudin, ART—123, Idraparinux, SR—123781, AZD—0837, MCC—977, TGN— 255, TGN—167, RWJ—58436, LB—30870, MPC—09 20, Pegmusirudin ゝ Org—426751, etc., thrombolytic drugs (eg, urokina se, tisokinase, anoleplase, alteplase) (natepla se), monteplase, pamiteplase, etc.), platelet aggregation inhibitors (eg, ticlepidine hydrochloride, sirosta) Zol (cilost azol), icosapentate ethinole, beraprost sodium, sanogre pogrelate hydrochloride, etc., Xa inhibitors (eg, Fondaparinux, BAY— 59— 7939, DU— 176b, YM—150, SR—126517, Apixaban, Raz axaban, LY—517717, MLN—102, Octaparine, Otamixaban, EMD—503 982, TC—10, CS—3030, AVE—3247, GSK—813893, KFA—1982, etc.), in plasma And carboxypeptidase B (also known as activated thrombin-activatable fibrinol ysis inhibitor [TAFIa]) inhibitors (eg, AZD-9684, EF-6265, MN-462).
[0086] 本発明の化合物を医薬として提供する場合、固形剤、液剤等の種々の態様の製剤 形態を適宜に採択することができる。その際、製薬学的に許容される担体を配合する ことも可能である。そのような担体の例としては、一般的な賦形剤、増量剤、結合剤、 崩壊剤、被覆剤、糖衣剤、 pH調整剤、溶解剤又は水性若しくは非水性溶媒などが 挙げられる。本発明の化合物とこれらの担体から、錠剤、丸剤、カプセル剤、顆粒剤 、粉剤、散剤、液剤、乳剤、懸濁剤、注射剤等を調製することができる。 [0086] When the compound of the present invention is provided as a medicament, various forms of preparations such as solid preparations and liquid preparations can be appropriately adopted. At that time, a pharmaceutically acceptable carrier can be blended. Examples of such carriers include common excipients, bulking agents, binders, disintegrants, coating agents, dragees, pH adjusters, solubilizers or aqueous or non-aqueous solvents. Tablets, pills, capsules, granules, powders, powders, solutions, emulsions, suspensions, injections and the like can be prepared from the compound of the present invention and these carriers.
[0087] また、本発明の化合物は、 a、 β若しくは γ—シクロデキストリン又はメチルイ匕シクロ デキストリン等に包接させて、その溶解性を改善することも可能である。
[0088] 本発明の化合物の投与量は、疾患、症状、体重、年齢、性別、投与経路等により異 なってくる力 S、成人に対し、 1曰当たり 0. 1〜: LOOOmg/kg体重であり、 0. 1〜200 mgZkg体重が好ましぐ 0. l〜10mgZkg体重がより好ましい。これを 1日 1回から 数回に分けて投与することができる。 [0087] The solubility of the compound of the present invention can also be improved by inclusion in a, β or γ-cyclodextrin, methylicyclodextrin, or the like. [0088] The dose of the compound of the present invention varies depending on the disease, symptom, body weight, age, sex, route of administration, etc. S, for adults, 0.1 to 1 per person: LOOOmg / kg body weight Yes, 0.1 to 200 mg Zkg body weight is preferred 0.1 to 10 mg Zkg body weight is more preferred. This can be administered once to several times a day.
実施例 Example
[0089] 以下に実施例及び試験例をあげて本発明をさらに詳しく説明するが、本発明はこ れらの記載によって限定的に解釈されるものではない。 [0089] Hereinafter, the present invention will be described in more detail with reference to Examples and Test Examples, but the present invention should not be construed as being limited by these descriptions.
[0090] 実施例 1 [0090] Example 1
4 - {4 - [2 - (ベンジルォキシ)エトキシ]—2—メチルベンジル } 5—イソプロピ ルー 1 H ピラゾール 3 ィル 5 チォ— β — Ό—ダルコビラノシドの合成 4-{4-[2- (Benzyloxy) ethoxy] -2-methylbenzyl} 5-isopropylidyl 1 H pyrazole 3 yl 5 thio- β — Ό-Dalcobilanoside synthesis
[0091] [化 9] [0091] [Chemical 9]

[0092] ( 1) 4— {4 [2 (ベンジルォキシ)エトキシ ] 2 メチルベンジル } 5 イソプロ ピル 1H ピラゾールー 3—ィル 2, 3, 4, 6—テトラー Ο ァセチルー 5 チォー β - D -ダルコビラノシドの合成 [0092] (1) 4— {4 [2 (Benzyloxy) ethoxy] 2 Methylbenzyl} 5 Isopropyl 1H Pyrazole-3-yl 2, 3, 4, 6-Tetra ァ Acetyl-5 Tio β-D-Darcobilanoside Synthesis
2, 3, 4, 6—テトラ一 Ο ァセチルー 5 チォ一 D—ダルコビラノース(0. 582g、 1 . 60mmol)、 4 { [4一(2 べンジルォキシエトキシ) 2 メチルフエ-ル]メチル } —1 , 2 ジヒドロ一 5—イソプロピル一 3H ピラゾール一 3—オン(0. 304g、 0. 79 9mmol;国際公開 WO04Z050122号を参考に 4 -ブロモ 3 メチルフエノールよ り合成)及びトリフエ-ルホスフィン(0. 420g、 1. 60mmol)のテトラヒドロフラン(lmL )溶液に氷冷下、ジイソプロピルァゾジカルボキシレート(0. 809mL、 1. 60mmol) の 40%トルエン溶液をゆっくりと滴下した。氷冷下で 30分間攪拌し、室温に昇温後、 さらに 3時間攪拌した。溶媒を減圧下留去し、得られた残渣をシリカゲルカラムクロマ トグラフィ一(n—へキサン:酢酸ェチル = 1 : 1)にて 2回精製した後、薄層クロマトダラ
α- ϋ
 ' 'ε 'ζ / — ε— /— ½ —2, 3, 4, 6-Tetra-l-acetylyl 5-thio-D-Dalcoviranose (0. 582g, 1.60mmol), 4 {[[4- (2-Benzyloxyethoxy) 2methylphenol] methyl} —1, 2, 2 Dihydro-1,5-isopropyl-1,3H Pyrazol-1,3-one (0.304 g, 0.79 9 mmol; synthesized from 4-bromo-3-methylphenol with reference to WO 04Z050122) and triphenylphosphine (0 To a solution of 420 g, 1.60 mmol) in tetrahydrofuran (lmL) was slowly added dropwise a 40% toluene solution of diisopropylazodicarboxylate (0.809 mL, 1.60 mmol) under ice cooling. The mixture was stirred for 30 minutes under ice-cooling, warmed to room temperature, and further stirred for 3 hours. The solvent was distilled off under reduced pressure, and the resulting residue was purified twice by silica gel column chromatography (n-hexane: ethyl acetate = 1: 1), and then thin-layer chromatographer. α- ϋ '' ε 'ζ / — ε— / — ½ —
' 'ε 'ζ / — ε— /— ½ —2, 3, 4, 6-Tetra-l-acetylyl 5-thio-D-Dalcoviranose (0. 582g, 1.60mmol), 4 {[[4- (2-Benzyloxyethoxy) 2methylphenol] methyl} —1, 2, 2 Dihydro-1,5-isopropyl-1,3H Pyrazol-1,3-one (0.304 g, 0.79 9 mmol; synthesized from 4-bromo-3-methylphenol with reference to WO 04Z050122) and triphenylphosphine (0 To a solution of 420 g, 1.60 mmol) in tetrahydrofuran (lmL) was slowly added dropwise a 40% toluene solution of diisopropylazodicarboxylate (0.809 mL, 1.60 mmol) under ice cooling. The mixture was stirred for 30 minutes under ice-cooling, warmed to room temperature, and further stirred for 3 hours. The solvent was distilled off under reduced pressure, and the resulting residue was purified twice by silica gel column chromatography (n-hexane: ethyl acetate = 1: 1), and then thin-layer chromatographer. α- ϋ '' ε 'ζ / — ε— / — ½ —
— ci i — g—[ べ > ^/^ ー (^i^^ ^-z) — Ci i — g— [be> ^ / ^ ー (^ i ^^ ^ -z)

[Οΐ^ ] -^ >Λ^ ^ / —a— ϋ— ^ -9 / -E- /— ½ — m— — g—[ べ > ^/^ ー (^i^^ ^-z) -^]→ ^ ^ ^ °^ ¾(%^6 §6so Ό)呦 挲ェつ; 、つ it エ^ (ε: z I = [Οΐ ^]-^> Λ ^ ^ / —a— ϋ— ^ -9 / -E- / — ½ — m— — g— [Be> ^ / ^ ー (^ i ^^ ^ -z)-^ ] → ^ ^ ^ ° ^ ¾ (% ^ 6 §6so Ό) 呦 挲 etsu; 、 tsu it e ^ (ε: z I =
爵止 教
 Baronism
Baronism
•(H S 'ω) ε ·Ζ - IZ'L (Η ΐ 'ZH SS"8=f 'Ρ) 6Γ9 (H ΐ 'ZH 6¥Ζ=ί 'Ρ) ZL'9 (H ΐ 'ZH 6VZ 'SS'8=f 'ΡΡ) ΐ9·9 (H ΐ '^Η 98"8=f 'P) 98'S (H ΐ '^Η 9 8"8=f ' ) IS'S (H ΐ 'ZH 6Z"6=f ' ) ZS"S (H ΐ '^Η 6Z"6=f ' ) ΖΓ3 (H Z 's) £9'f (H ΐ 'z• (HS 'ω) ε · Ζ-IZ'L (Η ΐ' ZH SS "8 = f 'Ρ) 6Γ9 (H ΐ' ZH 6 ¥ Ζ = ί 'Ρ) ZL'9 (H ΐ' ZH 6VZ 'SS'8 = f 'ΡΡ) ΐ9 · 9 (H ΐ' ^ Η 98 "8 = f 'P) 98'S (H ΐ' ^ Η 9 8" 8 = f ') IS'S (H ΐ' ZH 6Z "6 = f ') ZS "S (H ΐ' ^ Η 6Z" 6 = f ') ΖΓ3 (HZ' s ) £ 9'f (H ΐ 'z
H m 'ζ6·π=ί" 'ΡΡ) es^ (H ε svf - so' (H Z ssx - 9∑τ (H ε 6sx - 8ε·ε (H ΐ 98 -
 (Η ε (Η ε so (Η ε so (Η ε 86·ΐ (Η ε 's) 08· ΐ (Η 9 'ΖΗ 66·9=ί" 'Ρ) ZVI d g (ρ- Ο ΟΗΟΊΗ 'ζ 00S) Η匪 Ηΐ H m 'ζ6 · π = ί "' ΡΡ) es ^ (H ε svf-so '(HZ ssx-9∑τ (H ε 6sx-8ε · ε (H ΐ 98- (Η ε (Η ε so (Η ε so (Η ε 86 · ΐ (Η ε 's) 08 · ΐ (Η 9' Ζ · 66 9 = ί "'Ρ) ZVI dg (ρ- Ο ΟΗΟΊΗ' ζ 00S) Η 匪 Ηΐ
(Η ε (Η ε so (Η ε so (Η ε 86·ΐ (Η ε 's) 08· ΐ (Η 9 'ΖΗ 66·9=ί" 'Ρ) ZVI d g (ρ- Ο ΟΗΟΊΗ 'ζ 00S) Η匪 Ηΐ H m 'ζ6 · π = ί "' ΡΡ) es ^ (H ε svf-so '(HZ ssx-9∑τ (H ε 6sx-8ε · ε (H ΐ 98- (Η ε (Η ε so (Η ε so (Η ε 86 · ΐ (Η ε 's) 08 · ΐ (Η 9' Ζ · 66 9 = ί "'Ρ) ZVI dg (ρ- Ο ΟΗΟΊΗ' ζ 00S) Η 匪 Ηΐ
°- (%ι ^ Ο Ό)呦^ ^m ^ ^ ^Ww、つ攝慰エ^ (ΐ :os= 一, マ / °-(% ι ^ Ο Ό) 呦 ^ ^ m ^ ^ ^ Ww, congratulations ^ (ΐ: os = one, ma /
98C6S0/.00Zdf/X3d 93 8996ΖΪ/.00Ζ OAV
^ ^^( ^-\ (^S) /— ^0) (louiuiQ2,2 ·ο SQ I Ό)、 έ I= / ー α— ϋ
 'ρ 'ε 'ζ / —ε— /— ½ 98C6S0 / .00Zdf / X3d 93 8996ΖΪ / .00Ζ OAV ^ ^^ (^-\ (^ S) / — ^ 0) (louiuiQ2,2 · ο SQ I Ό), έ I = / ー α— ϋ 'ρ' ε 'ζ / —ε— / — ½
'ρ 'ε 'ζ / —ε— /— ½ 98C6S0 / .00Zdf / X3d 93 8996ΖΪ / .00Ζ OAV ^ ^^ (^-\ (^ S) / — ^ 0) (louiuiQ2,2 · ο SQ I Ό), έ I = / ー α— ϋ 'ρ' ε 'ζ / —ε— / — ½
— m— — g— [ べ > ^/^ ー (^i^^ ^-z) -^]→— M— — g— [Be> ^ / ^ ー (^ i ^^ ^ -z)-^] →
-^ >Λ^ ^ / —a— ϋ— ^ -9 / -E- /— ½ - m— ci i — g— [ べ > ^/^ ー (^i^^ ^-z)→~]→{z) -^> Λ ^ ^ / —a— ϋ— ^ -9 / -E- / — ½-m— ci i — g— [be> ^ / ^ ー (^ i ^^ ^ -z) → ~] → (z)
•(Η ΐ 'ΖΗ SS"8=f 'Ρ) ΐ • (Η ΐ 'ΖΗ SS "8 = f' Ρ) ΐ
8·9 (Η ΐ 'ΖΗ WZ=[ 'Ρ) ZL'9 (Η ΐ '^Η WZ 'SS'8=f 'ΡΡ) 29"9 (Η ΐ '^Η 98·8=ί" 'Ρ) 38 •S (Η ΐ 'ΖΗ 98·8=ί" 6V (Η ΐ '^Η U'6=f SS"S (Η ΐ '^Η U'6=f 9Γ3 (Η ΐ 'ζ8 · 9 (Η ΐ 'ΖΗ WZ = [' Ρ) ZL'9 (Η ΐ '^ Η WZ' SS'8 = f 'ΡΡ) 29 "9 (Η ΐ' ^ Η 98 · 8 = ί" 'Ρ ) 38 • S (Η ΐ 'ΖΗ 98 · 8 = ί "6V (Η ΐ' ^ Η U'6 = f SS" S (Η ΐ '^ Η U'6 = f 9Γ3 (Η ΐ' ζ
Η 6s' '68·π=ί" 'ΡΡ) εε· (Η ε svf - ββτ (Η Ζ Ζ6·ε - 68·ε (Η ε 。9·ε - ζε·ε (Η ΐ 68 - ζυζ (Η ε ιτζ (Η ε so (Η ε so (Η ε 86·ΐ (Η ε 's) 28"ΐ (Η 9 'ΖΗ 66·9=ί" 'Ρ) fVl d g (p- Ο ΟΗΟΊΗ 'ζ 00S) 匪 Ηΐ
 Η 6s ''68 · π = ί "' ΡΡ) εε · (Η ε svf-ββτ (Η Ζ Ζ6 · ε-68 · ε (Η ε .9 · ε-ζε · ε (Η ΐ 68-ζυζ (Η ε ιτζ (Η ε so (Η ε so (Η ε 86 · ΐ (Η ε 's) 28 "ΐ (Η 9' Ζ Η 66 9 = ί"'Ρ) fVl dg (p- Ο ΟΗΟΊΗ' ζ 00S ) 匪 Ηΐ
Η 6s ''68 · π = ί "' ΡΡ) εε · (Η ε svf-ββτ (Η Ζ Ζ6 · ε-68 · ε (Η ε .9 · ε-ζε · ε (Η ΐ 68-ζυζ (Η ε ιτζ (Η ε so (Η ε so (Η ε 86 · ΐ (Η ε 's) 28 "ΐ (Η 9' Ζ Η 66 9 = ί"'Ρ) fVl dg (p- Ο ΟΗΟΊΗ' ζ 00S ) 匪 Ηΐ
- ^ 止 ^蹈 峯氺 ^w ^z ·ου峯 —マ ^ ^難氺
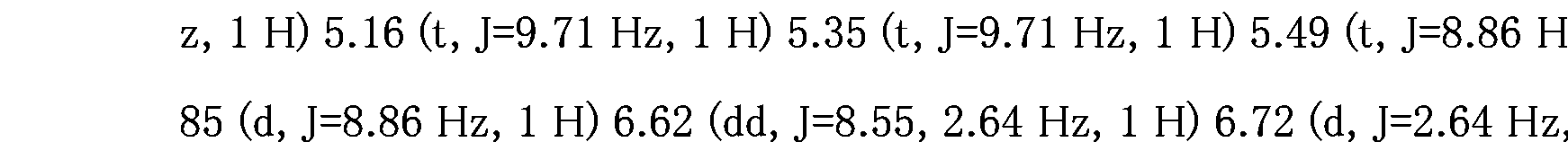 -^ Stop ^ 蹈 峯 氺 ^ w ^ z · ου 峯 —Ma ^^
-^ Stop ^ 蹈 峯 氺 ^ w ^ z · ου 峯 —Ma ^^
M ^m^、つ ¾^ οε、 止^^ m止縱; C)> Φ¾ 缀べエ AI。/O
 继缀
继缀
 §8 ·9ε)ベ / ェ fH、M ^ m ^, ¾ ^ οε, Stop ^^ m Stop; C)> Φ¾ 缀 Bayer AI. / O 继 缀 §8
§8 ·9ε)ベ / ェ fH、M ^ m ^, ¾ ^ οε, Stop ^^ m Stop; C)> Φ¾ 缀 Bayer AI. / O 继 缀 §8
) Τ 一,ェ ^ ε rd : ^ % ¾ssx oso/ oo ^国: \om ) Junichi ^ ε rd: ^% ssx oso / oo ^ Country: \ om
ΌΖ § · 92 ) ^-^ - S - — .6c¾ - HS - - 9 - - Ζ Ί- {
 -Ζ) -V] }→ (louiuix · 0Ζ §1 ·ΐ9) 一 r= / — α ^ S ^^ — Ο— έ4 — 9 ' '£ 'Ζ § § · 92) ^-^-S-— .6c¾-HS--9--Ζ Ί- { -Ζ) -V]} → (louiuix · 0Ζ § 1 · ΐ9) One r = / — α ^ S ^^ — Ο— έ4 — 9 '' £ 'Ζ
-Ζ) -V] }→ (louiuix · 0Ζ §1 ·ΐ9) 一 r= / — α ^ S ^^ — Ο— έ4 — 9 ' '£ 'Ζ § § · 92) ^-^-S-— .6c¾-HS--9--Ζ Ί- { -Ζ) -V]} → (louiuix · 0Ζ § 1 · ΐ9) One r = / — α ^ S ^^ — Ο— έ4 — 9 '' £ 'Ζ
98C6S0/.00Zdf/X3d 93 8996ΖΪ/.00Ζ OAV
ェチル:エタノール:水 = 20 : 2 : 1〜 15 : 2 : 1)にて精製し、白色粉末として表題化合 物 (0. 106g、 82%)を得た。得られた化合物の構造、 NMRデータ及び MSデータ を表 1に示す。 98C6S0 / .00Zdf / X3d 93 8996ΖΪ / .00Ζ OAV Ethyl: ethanol: water = 20: 2: 1 to 15: 2: 1) to obtain the title compound (0.106 g, 82%) as a white powder. The structure, NMR data and MS data of the obtained compound are shown in Table 1.
[0095] 実施例 3 [0095] Example 3
ί アミノエトキシ) 2—メチルベンジル]—5—イソプロピル ί aminoethoxy) 2-methylbenzyl] -5-isopropyl
ィル 5—チォー j8— D—ダルコビラノシドの合成 Yyl 5-thio j8—D—Synthesis of darcobilanoside
[0096] [化 11] [0096] [Chemical 11]

[0097] (1) 4— [4— (2 アジドエトキシ) 2—メチルベンジル」 5—イソプロピル一 1H— ピラゾールー 3—ィル 2, 3, 4, 6—テトラー O ァセチルー 5 チォー 13—D—グ ルコビラノシドの合成 [0097] (1) 4— [4— (2 Azidoethoxy) 2-methylbenzyl ”5-Isopropyl 1H-pyrazole-3-yl 2, 3, 4, 6-tetra-O-acetylyl 5-thio 13-D-g Synthesis of lucobilanoside
実施例 2 (1)で合成した 4— [4— (2 ヒドロキシエトキシ) 2—メチルベンジル] - 5—イソプロピル一 1H ピラゾール一 3—ィル 2, 3, 4, 6—テトラ一 O ァセチル —5 チォ一 13—D—ダルコビラノシド(8. 38g、 13. 2mmol)、トリエチルァミン(2. 8mL、 20mmol)のクロ口ホルム(82mL)溶液に氷冷下、メタンスルホユルクロリド(1 . 2mL、 13mmol)をカ卩え、室温で 30分間攪拌した。反応液に 0. 5N塩酸水溶液を 加え、酢酸ェチルで抽出後、有機層を飽和重曹水溶液、飽和食塩水で洗浄し、硫酸 マグネシウムで乾燥した。乾燥剤をろ別後、溶媒を減圧下留去した。残渣を N, N— ジメチルホルムアミドに溶解させアジィ匕ナトリウムをカ卩えて 100°Cで 3時間攪拌した。 反応液に水を加え、酢酸ェチルで 2回抽出した後、有機層を飽和食塩水で 3回洗浄 し、硫酸マグネシウムで乾燥した。乾燥剤をろ別後、溶媒を減圧下留去し、残渣をシ リカゲルカラムクロマトグラフィ—(n—へキサン:酢酸ェチル = 1 : 1)にて精製し、淡黄 色粉末として表題ィ匕合物(6. 98g、 80%)を得た。 Example 2 4- [4- (2 Hydroxyethoxy) 2-methylbenzyl] -5-isopropyl 1H 1 Pyrazole 3-yl 2, 3, 4, 6-Tetra 1 O acetyl 3 synthesized in (1) 13-D-Dalcobilanoside (8. 38 g, 13.2 mmol) and triethylamine (2.8 mL, 20 mmol) in chlorosulfur chloride (1.2 mL, 13 mmol) under ice-cooling were added to a chloroform solution (82 mL). ) And stirred at room temperature for 30 minutes. A 0.5N aqueous hydrochloric acid solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with a saturated aqueous sodium bicarbonate solution and saturated brine, and dried over magnesium sulfate. After the desiccant was filtered off, the solvent was distilled off under reduced pressure. The residue was dissolved in N, N-dimethylformamide, sodium azide was added, and the mixture was stirred at 100 ° C. for 3 hours. Water was added to the reaction mixture, and the mixture was extracted twice with ethyl acetate. The organic layer was washed 3 times with saturated brine and dried over magnesium sulfate. After filtering off the desiccant, the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography (n-hexane: ethyl acetate = 1: 1) to give the title compound as a pale yellow powder. (6.98 g, 80%) was obtained.
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.14 (d, J=6.99 Hz, 6 H) 1.82 (s, 3 H) 1.98 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.28 (s, 3 H) 2.76 - 2.89 (m, 1 H) 3.35
 1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.14 (d, J = 6.99 Hz, 6 H) 1.82 (s, 3 H) 1.98 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.28 (s, 3 H) 2.76-2.89 (m, 1 H) 3.35
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.14 (d, J = 6.99 Hz, 6 H) 1.82 (s, 3 H) 1.98 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.28 (s, 3 H) 2.76-2.89 (m, 1 H) 3.35
^£ a) u ^£ -& (z)z m 、ェ、 ∑= / ー a— g ^ £ a) u ^ £-& (z) z m, e, ∑ = / ー a— g
' 'ε 'z / — ε— /— ½ — m— ΰ : '' ε 'z / — ε— / — ½ — m— ΰ:
( 、 ェ,^ 一
 (, E, ^ one
(, E, ^ one
ー α— ϋ
 'ρ 'ε 'ζ / —ε— /— ½ — m— — g—[ べ > ^/^ ー (^i^^ ^-z) -^]→ー α— ϋ 'ρ' ε 'ζ / —ε— / — ½ — m— — g— [Be> ^ / ^ ー (^ i ^^ ^ -z)-^] →
'ρ 'ε 'ζ / —ε— /— ½ — m— — g—[ べ > ^/^ ー (^i^^ ^-z) -^]→ー α— ϋ 'ρ' ε 'ζ / —ε— / — ½ — m— — g— [Be> ^ / ^ ー (^ i ^^ ^ -z)-^] →
-^ >Λ^ ^ / -α- ϋ - ^ -9 / -E- /— ½ — m— 一 g—[ べ > ^/^ ー ( ^ ェ,^ 一 ]— (ε) -^> Λ ^ ^ / -α- ϋ-^ -9 / -E- / — ½ — m— one g— [be> ^ / ^ ー (^ é, ^ one] — (ε)
•(Η ΐ 'ZH 6S"8=f 'Ρ) 08·9• (Η ΐ 'ZH 6S "8 = f' Ρ) 08 · 9
(Η ΐ 'ZH WZ=[ 'Ρ) U'9 (Η ΐ 'ZH WZ '6S"8=f 'ΡΡ) 09·9 (Η ΐ '^Η 98·8=ί" 'Ρ) S8"S ( Η ΐ 'ΖΗ 98·8=ί" ' ) OS'S (Η ΐ '^Η 6Z"6=f ') 9S"S (Η ΐ '^Η 6Z"6=f ' ) 9Γ3 (Η ΐ '^Η 9(Η ΐ 'ZH WZ = [' Ρ) U'9 (Η ΐ 'ZH WZ' 6S "8 = f 'ΡΡ) 09 · 9 (Η ΐ' ^ Η 98 · 8 = ί" 'Ρ) S8 "S (Η ΐ 'ΖΗ 98 · 8 = ί "') OS'S (Η ΐ '^ Η 6Z" 6 = f') 9S "S (Η ΐ '^ Η 6Z" 6 = f') 9Γ3 (Η ΐ '^ Η 9
9· ' 6"n=f 'ΡΡ) wf (Η ε 'ω) oz-f - 98·ε (Η ε 69·ε - ητ (Η Ζ 'ΖΗ sre=f ') οτ (Η ΐ wz - sz (Η ε ιτζ (Η ε so (Η ε ζο~ζ (Η ε 86·ΐ (Η ε 's) Ι8·ΐ (Η 9 'ΖΗ 66·9=ί" 'Ρ) 2VI d g (p- Ο ΟΗΟΊΗ 'ζ 00S) 匪 Ηΐ 9 '6 "n = f' ΡΡ) wf (Η ε 'ω) oz-f-98 · ε (Η ε 69 · ε-ητ (Η Ζ' Ζ sre = f ') οτ (Η Η wz- sz (Η ε ιτζ (Η ε so (Η ε ζο ~ ζ (Η ε 86 · ΐ (Η ε 's) Ι8 · ΐ (Η 9' Ζ 66 · 9 = ί "'Ρ) 2VI dg (p- ΟΗΟΊΗ ' ζ 00S) 匪 Ηΐ
¾(%i6 §9i -9)
 : v
: v
 、つ辛爵止 S教 继 9 ¾ (% i6 §9i -9) : V , Spicy Suicide S Teaching 9
、つ辛爵止 S教 继 9 ¾ (% i6 §9i -9) : V , Spicy Suicide S Teaching 9
)
 )
)
/ ー a— ϋ
 ' 'ε 'ζ / ε— /— 、έ / ー a— ϋ '' ε 'ζ / ε— / —, έ
' 'ε 'ζ / ε— /— 、έ / ー a— ϋ '' ε 'ζ / ε— / —, έ
0¾ - HI - - 9 -
 一 ー a— ϋ
一 ー a— ϋ
 'ε 'ζ / —ε— /— ½ — m— 一 g—[ べ > ^/^ ー ( ^ ェ,^ 一 →~]→{z) 0 ¾-HI--9- One ー a— ϋ 'ε' ζ / —ε— / — ½ — m— One g— [Be> ^ / ^ ー (^ ́, ^ One → ~] → {z)
'ε 'ζ / —ε— /— ½ — m— 一 g—[ べ > ^/^ ー ( ^ ェ,^ 一 →~]→{z) 0 ¾-HI--9- One ー a— ϋ 'ε' ζ / —ε— / — ½ — m— One g— [Be> ^ / ^ ー (^ ́, ^ One → ~] → {z)
·( · (
H ΐ 'ZH SS"8=f 'Ρ) ΐ8·9 (Η ΐ '^Η WZ=[ 'Ρ) ZL'9 (Η ΐ '^Η 9 'SS"8=f 'ΡΡ) ΐ9·9 (Η ΐ 'ΖΗ 98·8=ί" 'Ρ) S8"S (Η ΐ '^Η 98·8=ί" ') OS'S (Η ΐ '^Η 6Z"6=f ' ) 9S"S (Η ΐ '^Η 6 ζ·6=ί" ') ere (Η Ϊ '^Η 99· ' 6·π=ί" 'ΡΡ) ~f (Η ε svf - eo^ (Η e ε9·ε - H ΐ 'ZH SS "8 = f' Ρ) ΐ8 · 9 (Η ΐ '^ Η WZ = [' Ρ) ZL'9 (Η ΐ '^ Η 9' SS" 8 = f 'ΡΡ) ΐ9 · 9 ( Η ΐ 'ΖΗ 98 · 8 = ί "' Ρ) S8" S (Η ΐ '^ Η 98 · 8 = ί "') OS'S (Η ΐ '^ Η 6Z" 6 = f') 9S "S (Η ΐ '^ Η 6 ζ · 6 = ί "') ere (Η Ϊ '^ Η 99 ·' 6 · π = ί" 'ΡΡ) ~ f (Η ε svf-eo ^ (Η e ε9 · ε-
98C6S0/.00Zdf/X3d 83 8996ΖΪ/.00Ζ OAV
Γ8 (Η Οΐ WL (Η ΐ 'ΖΗ 6ε·8=ί" 'Ρ) 6Γ9 (Η ΐ '^Η WZ=[ 'Ρ) U'9 (Η ΐ '^Η WZ 98C6S0 / .00Zdf / X3d 83 8996ΖΪ / .00Ζ OAV Γ8 (Η Οΐ WL (Η ΐ 'ΖΗ 6ε · 8 = ί "' Ρ) 6Γ9 (Η ΐ '^ Η WZ = [' Ρ) U'9 (Η ΐ '^ Η WZ
'6ε·8=ί" 'ΡΡ) 6S.9 (Η ΐ 'ΖΗ 98·8=ί" 'Ρ) ZS' (Η ΐ '^Η 98·8=ί" ') 6V (Η ΐ '^Η 6Z"6=f ') 9S"S (Η S 'ω) - ZO"S (Η ΐ '^Η 06· '68·ΐΐ=ί" 'ΡΡ) Γ (Η S 'ω) 8ΐ· - 66·ε '6ε · 8 = ί "' ΡΡ) 6S.9 (Η ΐ 'ΖΗ 98 · 8 = ί"' Ρ) ZS '(Η ΐ' ^ Η 98 · 8 = ί "') 6V (Η ΐ' ^ Η 6Z "6 = f ') 9S" S (Η S' ω)-ZO "S (Η ΐ '^ Η 06 · '68 · ΐΐ = ί"' ΡΡ) Γ (Η S 'ω) 8ΐ ·-66 · ε
(Η ζ 98·ε - τ (Η ζ ΐ9·ε - ·ε (Η Ϊ 'ω) βζτ - οε·ε (Η Ϊ - 2 (Η ε 's) ιτζ (Η ε so (Η ε ζο~ζ (Η ε Ζ6·ΐ (Η ε ΐ8·ΐ (Η ε 'ΖΗ ε9·ΐ= f 'Ρ) 3Γΐ (Η S 'ΖΗ ε9·ΐ=ί" 'Ρ) ZVl radd 9 '^Η 00S) Η Ν Ηΐ (Η ζ 98 · ε-τ (Η ζ ΐ9 · ε-· ε (Η Ϊ 'ω) βζτ-οεε (Η Ϊ-2 (Η ε' s) ιτζ ( (Η ε Ζ6 · ΐ (Η ε ΐ8 · ΐ (Η ε 'ΖΗ ε9 · ΐ = f' Ρ) 3Γΐ (Η S 'ΖΗ ε9 Ηΐ
(%εζ ¾½ Ό) <¾?^®¾^α^¾¾^^、つ攝慰エ^ (I: I = ェ邈 : ベ ^ —u) 、つ辛爵 止 s教 (% εζ ¾½ Ό) <¾? ^ ®¾ ^ α ^ ¾¾ ^^, 攝 攝 comfort e ^ (I: I = 邈: ^ ^ —u)
M 。
 ベ ^:^ M. Be ^: ^
ベ ^:^ M. Be ^: ^
- 1— /— ½ — Ηΐ - ( ^ / ^^ べ:^) ^- {N、N ^ 缀 (Ύ^£ · -1— / — ½ — Ηΐ-(^ / ^^ be: ^) ^- { N, N ^ 缀 (Ύ ^ £ ·
0)^6^^¾ ^O) (10UIUIX6 Ό §S09 Ό) ^?^ ( ^ - α - ϋ -
 'ε 'ζ / 一 ε— /— ½ 一 m— ΰ : (0) ^ 6 ^^ ¾ ^ O) (10UIUIX6 § § S09 Ό) ^? ^ (^-Α-ϋ- 'ε' ζ / One ε— / — ½ m— ΰ:
'ε 'ζ / 一 ε— /— ½ 一 m— ΰ : (0) ^ 6 ^^ ¾ ^ O) (10UIUIX6 § § S09 Ό) ^? ^ (^-Α-ϋ- 'ε' ζ / One ε— / — ½ m— ΰ:
- 9 - \Λ(^ ^Λ(^-Ζ - ( ^^ェ, — -^]→-^ ^ (z) w , ム &^^ ( ^ { ^エ ェ ^ — ε - ( ^ { / — — 一 m— [ ^^ ( / έ r= / — α— ϋ— ^— s— — ο -9-\ Λ (^ ^ Λ (^-Ζ-(^^ e, —-^] →-^ ^ (z) w, Mu & ^^ (^ {^ et ^ — ε-(^ {/ — — One m— [^^ (/ έ r = / — α— ϋ— ^ — s— — ο

[Zl^ [6600] [Zl ^ [6600]
—ム [ ^ ( ^ { ^ ェ [ ^,ェ ^ — ε— ( ^ { / — — /— ½ — m— [ ^^ ( / έ —M [^ (^ {^ é [^, é ^ — ε— (^ {/ — — / — ½ — m— [^^ (/ έ
に / 一 α— ϋ ]
 Ni / a α- ϋ]
Ni / a α- ϋ]
ϋ¾Ϊ [8600] ϋ¾Ϊ [8600]
。 · . ·
98C6S0/.00Zdf/X3d 63 8996ΖΪ/.00Ζ OAV
2 (t, J=5.28 Hz, 1 H) 11.72 (br. s., 1 H). 98C6S0 / .00Zdf / X3d 63 8996ΖΪ / .00Ζ OAV 2 (t, J = 5.28 Hz, 1 H) 11.72 (br. S., 1 H).
(2)ベンジル [ィミノ({ 2— [4—( 一イソプロピル一 3— [ (5 チォ一 β— D グノレ コピラノシル)ォキシ]— 1H ピラゾール一 4—ィル }メチル) 3—メチルフエノキシ] ェチル }ァミノ)メチル]力ルバマートの合成 (2) Benzyl [imino ({2- [4 -— (Isopropyl-1-3-((5-thiol-β-D-gnolecopyranosyl) oxy]-1H-pyrazole-4-yl} methyl) 3-methylphenoxy] ethyl} amino ) Synthesis of methyl] force rubamate
ジベンジル [ (E)— ( { 2— [4—( 一イソプロピル一 3— [ (2, 3, 4, 6—テトラ一 Ο —ァセチル一 5 チォ一 13—D—ダルコピラノシル)ォキシ ] 1Η ピラゾールー 4 ーィル }メチル) 3—メチルフエノキシ]ェチル }ァミノ)メチルイリデン]ビス力ルバマ ート(0. 530g、0. 560mmol)のべンジルアルコール(lmL)溶液にナトリウムベンジ ルォキシドのべンジルアルコール溶液(1M、 0. 34mL、 0. 34mmol)を室温でカロえ 6時間攪拌した。反応液に水を加え、酢酸ェチルで抽出後、硫酸マグネシウムで乾 燥した。乾燥剤をろ別後、溶媒を減圧下留去し、得られた残渣をシリカゲルカラムクロ マトグラフィ―(酢酸ェチル:エタノール:水 = 20: 2: 1)にて精製し、淡茶色粉末とし て表題化合物(0. 163g、 45%)を得た。得られたィ匕合物の構造、 NMRデータ及び MSデータを表 1に示す。 Dibenzyl [(E) — ({2— [4— (1 Isopropyl 1 3 — [(2, 3, 4, 6—Tetra 1 Ο —Acetyl 1 5 Thio 13—D-Darkopyranosyl) oxy] 1 Η Pyrazole 4 yl } Methyl) 3-methylphenoxy] ethyl} amino) methylylidene] bis-force rubamate (0.530 g, 0.560 mmol) in benzyl alcohol (lmL) solution with sodium benzyloxide in benzyl alcohol solution (1M, 0 34 mL, 0.34 mmol) was stirred at room temperature and stirred for 6 hours. Water was added to the reaction solution, extracted with ethyl acetate, and dried over magnesium sulfate. After filtering off the desiccant, the solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography (ethyl acetate: ethanol: water = 20: 2: 1) to give the title as a light brown powder. The compound (0.163 g, 45%) was obtained. Table 1 shows the structure, NMR data and MS data of the obtained compound.
[0101] 実施例 5 [0101] Example 5
N- { 2 - [4 - ( { 5 -イソプロピル 3— [ ( 5 チォ— β — Ό—ダルコピラノシル)ォ キシ] 1H ピラゾール— 4—ィル }メチル)—3—メチルフエノキシ]ェチル }グァ- ジンの合成 N- {2-[4-({5 -Isopropyl 3— [(5 thio- β — Ό-Dalcopyranosyl) oxy] 1H pyrazole-4-yl} methyl) -3-methylphenoxy] ethyl} guanidine Composition
[0102] [化 13] [0102] [Chemical 13]

[0103] 実施例 4で合成したベンジル [ィミノ({ 2—[4—( 一イソプロピルー3—[ (5 チ ォ一 13—D—ダルコピラノシル)ォキシ ] 1Η ピラゾール一 4—ィル }メチル) 3— メチルフエノキシ]ェチル }ァミノ)メチル]力ルバマート(0. 154g、0. 239mmol)のメ タノール(3mL)溶液に 20%水酸化パラジウム—活性炭素(0. 03g)を加え、水素雰 囲気下、室温にて 2時間攪拌した。不溶物をセライト濾過後、ろ液を減圧下留去し、
白色粉末として表題ィ匕合物(0. 108g、 85%)を得た。得られたィ匕合物の構造、 NM Rデータ及び MSデータを表 1に示す。 [0103] Benzyl synthesized in Example 4 [Imino ({2- [4 -— (Isopropyl-3-[(5 thio 13-D-Dalcopyranosyl) oxy] 1 1-pyrazole -1-yl} methyl) 3] —Methylphenoxy] ethyl} amino) methyl] powered rubamate (0.154 g, 0.239 mmol) in methanol (3 mL) was added with 20% palladium hydroxide-activated carbon (0.03 g), and at room temperature in a hydrogen atmosphere. For 2 hours. The insoluble material was filtered through Celite, and the filtrate was evaporated under reduced pressure. The title compound (0.108 g, 85%) was obtained as a white powder. Table 1 shows the structure, NMR data and MS data of the compound.
[0104] 実施例 6 [0104] Example 6
4 {4- [2- (N—力ルバモイルメチルァミノ)エトキシ ] 2 メチルベンジル } 5 —イソプロピル一 1H ピラゾール一 3—ィル 5 チォ一 13—D—ダルコビラノシドの 合成 4 {4- [2- (N-force ruberamoylmethylamino) ethoxy] 2 methylbenzyl} 5 —Isopropyl 1 H Synthesis of pyrazole 1 3-yl 5 thio 1 13-D-Dalcobilanoside
[0105] [化 14] [0105] [Chemical 14]

(1) 4— {4 [2—(N—力ルバモイルメチルァミノ)エトキシ ] 2 メチルベンジル } 5 イソプロピノレー 1H ピラゾーノレー3—ィノレ 2, 3, 4, 6—テトラー O ァセチ ルー 5—チォー 13 D—ダルコビラノシドの合成 (1) 4— {4 [2— (N-Strengthylamoylmethylamino) ethoxy] 2 Methylbenzyl} 5 Isopropynole 1H Pyrazonole 3-Inole 2, 3, 4, 6-Tetra O acetylene 5—Cheo 13 D—Synthesis of darcobilanoside
実施例 3 (2)で合成した 4— [4— (2 アミノエトキシ) 2—メチルベンジル]—5— イソプロピル一 1H ピラゾール一 3—ィル 2, 3, 4, 6—テトラ一 O ァセチル一 5 —チォ一 13—D—ダルコビラノシド(0. 507g、 0. 797mmol)の N、 N ジメチルホ ルムアミド(1. 6mL)溶液に 2 クロロアセトアミド(0. 112g、 1. 20mmol)、トリェチ ルァミン(0. 17mL、 1. 2mmol)をカ卩え、 50°Cで 3時間攪拌した。反応液をシリカゲ ルクロマトグラフィー(クロ口ホルム:メタノール =60 : 1)に付し、淡黄色粉末として表 題化合物(0. 185g、 34%)を得た。 Example 3 4- [4- (2 aminoethoxy) 2-methylbenzyl] -5-isopropyl 1H 1 pyrazole 1-3-yl 2, 3, 4, 6-tetra 1 O acetyl 1 synthesized in (2) 5 —Cho 13—D—To a solution of darcobilanoside (0.507 g, 0.797 mmol) in N, N dimethylformamide (1.6 mL), 2 chloroacetamide (0.112 g, 1.20 mmol), triethylamine (0.17 mL, 1. 2 mmol) was added and stirred at 50 ° C for 3 hours. The reaction solution was subjected to silica gel chromatography (black mouth form: methanol = 60: 1) to obtain the title compound (0.185 g, 34%) as a pale yellow powder.
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.13 (d, J=6.99 Hz, 6 H) 1.82 (s, 3 H) 1.97 (s, 3 H) 2.01 (s, 3 H) 2.03 (s, 3 H) 2.26 (s, 3 H) 2.71 - 2.89 (m, 1 H) 2.99 (t, J=4.66 Hz, 2 H) 3.30 - 3.59 (m, 5 H) 3.97 - 4.17 (m, 3 H) 4.30 (dd, J=11.97, 4.6 6 Hz, 1 H) 5.15 (t, J=9.64 Hz, 1 H) 5.34 (t, J=9.64 Hz, 1 H) 5.47 (t, J=8.78 Hz, 1 H ) 5.84 (d, J=8.78 Hz, 1 H) 6.30 - 6.45 (m, 1 H) 6.58 (dd, J=8.39, 2.41 Hz, 1 H) 6.68 (d, J=2.41 Hz, 1 H) 6.80 (d, J=8.39 Hz, 1 H) 7.18 - 7.30 (m, 1 H). 1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.13 (d, J = 6.99 Hz, 6 H) 1.82 (s, 3 H) 1.97 (s, 3 H) 2.01 (s, 3 H) 2.03 (s, 3 H) 2.26 (s, 3 H) 2.71-2.89 (m, 1 H) 2.99 (t, J = 4.66 Hz, 2 H) 3.30-3.59 (m, 5 H) 3.97-4.17 (m, 3 H) 4.30 ( dd, J = 11.97, 4.6 6 Hz, 1 H) 5.15 (t, J = 9.64 Hz, 1 H) 5.34 (t, J = 9.64 Hz, 1 H) 5.47 (t, J = 8.78 Hz, 1 H) 5.84 (d, J = 8.78 Hz, 1 H) 6.30-6.45 (m, 1 H) 6.58 (dd, J = 8.39, 2.41 Hz, 1 H) 6.68 (d, J = 2.41 Hz, 1 H) 6.80 (d, J = 8.39 Hz, 1 H) 7.18-7.30 (m, 1 H).
(2) 4— {4— [2—(N—力ルバモイルメチルァミノ)エトキシ ] 2 メチルベンジル }
—5—イソプロピノレ一 1H ピラゾーノレ一 3—ィノレ 5 チォ一 β—D—グノレコピラノシ ドの合成 (2) 4— {4— [2— (N-force ruberamoylmethylamino) ethoxy] 2 methylbenzyl} —5—Isopropinole 1H pyrazonole 1 3-inole 5 thio 1 β-D-Gnolecopyranosides
4— [4— (2 ヒドロキシエトキシ) 2—メチルベンジル]—5—イソプロピル一 1H— ピラゾールー 3—ィル 2, 3, 4, 6—テトラー Ο ァセチルー 5 チォー 13—D—グ ルコビラノシドの代わりに 4 {4- [2- (Ν—力ルバモイルメチルァミノ)エトキシ ] 2 —メチルベンジル } 5—イソプロピル一 1H ピラゾールー 3—ィル 2, 3, 4, 6—テ トラ一 Ο ァセチノレ一 5 チォ一 /3—D—ダルコビラノシドを用いて、実施例 2 (2)に 示す方法と同様の方法で表題化合物を合成した。得られた化合物の構造、 NMRデ ータ及び MSデータを表 1に示す。 4— [4— (2 Hydroxyethoxy) 2-methylbenzyl] —5-Isopropyl 1H—Pyrazol-3-yl 2, 3, 4, 6—Tetra ァ Acetyl 5 Thio 13—D—Instead of Glucobilanoside 4 {4- [2- (Ν—Force rubermoylmethylamino) ethoxy] 2 —Methylbenzyl} 5—Isopropylone 1H Pyrazol-3-yl 2, 3, 4, 6—Tetra 1 セ Acetinole 1 5 Thio 1 The title compound was synthesized in the same manner as shown in Example 2 (2) using / 3-D-darcobilanoside. Table 1 shows the structure, NMR data and MS data of the compound obtained.
[0107] 実施例 7 [0107] Example 7
N— (2—ヒドロキシ一 1, 1—ジメチノレエチノレ) N, 一 N— (2-Hydroxy 1 1, 1-Dimethyloletinole) N, 1
—3— [ (5—チォ一 13—D—ダルコピラノシル)ォキシ] —3— [(5-Chi 13-D-Darcopyranosyl) oxy]
}メチル) 3—メチルフエノキシ]ェチル }ゥレアの合成 } Methyl) 3-methylphenoxy] ethyl
[0108] [化 15] [0108] [Chemical 15]

[0109] 実施例 3 (2)で合成した 4— [4— (2 アミノエトキシ) 2—メチルベンジル]—5— イソプロピル一 1H ピラゾール一 3—ィル 2, 3, 4, 6—テトラ一 O ァセチル一 5 —チォ j8— D—ダルコビラノシド(0. 145g、 0. 228mmol)のクロ口ホルム(1. 5m L)溶液にトリェチルミン(0. 043mL、 0. 308mmol)、クロロギ酸 4 二トロフエ-ル (0. 053g、 0. 262mmol)を加え、室温で 1時間攪拌した。さらにトリェチルァミン(0 . 064mL、 0. 456mmol)と 2 アミノー 2—メチノレ一 1—プロノ ノーノレ(0. 033mL、 0. 342mmol)を加え、室温で 1時間攪拌した。溶媒を減圧下留去し、得られた残渣 にメタノール(2. 3mL)とナトリウムメトキシド(25%メタノール溶液、 0. 20mL、 0. 91 2mmol)を加え、室温で 4時間攪拌した。反応液にドライアイスをカ卩え、中和した後に 、溶媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸ェチ
— m— — g—[ べ > ^/^ ー ( ^ エ ^ pd、 — -v -v 一 α— ϋ / 一 ε— /— ½ 一 m— 一 g—[ / ベ:^ Example 3 4- [4- (2 aminoethoxy) 2-methylbenzyl] -5-isopropyl 1H synthesized pyrazole 1-3-yl 2, 3, 4, 6-tetra 1 O synthesized in (2) Acetyl 5 -thio j8- D-Dalcoviranoside (0.145 g, 0.228 mmol) in chloroform (1.5 mL) solution with triethylmine (0.043 mL, 0.308 mmol), chloroformate 4 ditrophenol ( 0.053 g, 0.262 mmol) was added, and the mixture was stirred at room temperature for 1 hour. Further, triethylamine (0.064 mL, 0.456 mmol) and 2-amino-2-methylol-1-prononorole (0.033 mL, 0.342 mmol) were added, and the mixture was stirred at room temperature for 1 hour. The solvent was evaporated under reduced pressure, and methanol (2.3 mL) and sodium methoxide (25% methanol solution, 0.20 mL, 0.991 2 mmol) were added to the resulting residue, and the mixture was stirred at room temperature for 4 hours. After dry ice was added to the reaction solution and neutralized, the solvent was distilled off under reduced pressure. The obtained residue was subjected to silica gel column chromatography (ethyl acetate — M— — g— [Be> ^ / ^ ー (^ d ^ pd, — -v -v One α— ϋ / One ε— / — ½ One m— One g— [/
•(H e se- - zz'L (H I 'ZH 6ε·8 • (H e se--zz'L (H I 'ZH 6ε
=f 'P) ZZ-9 (H ΐ 'ZH 6VZ=i 'Ρ) 99·9 (H ΐ '^Η 6VZ '6S"8=f 'PP) SS'9 (H ΐ '^Η 98·8= f 'P) S8"S (H ΐ 'ZH 98"8=f OS'S (H ΐ 'ω) \yq― OS'S (H ΐ '^Η ZF6=f SFS (H = f 'P) ZZ-9 (H ΐ' ZH 6VZ = i 'Ρ) 99 ・ 9 (H ΐ' ^ Η 6VZ '6S "8 = f' PP) SS'9 (H ΐ '^ Η 98.8 = f 'P) S8 "S (H ΐ' ZH 98" 8 = f OS'S (H ΐ 'ω) \ yq― OS'S (H ΐ' ^ Η ZF6 = f SFS (H
S <S) IS' (H ΐ 'ZH 99·, '68·ΐΐ=ί" 'ΡΡ) Wf (H ΐ '^Η ΐ8·ε '68·ΐΐ=ί" 'ΡΡ) ZVf (H Z <z S <S ) IS '(H ΐ' ZH 99 ·, '68 · ΐΐ = ί "'ΡΡ) Wf (H ΐ' ^ Η ΐ8 · ε '68 · ΐΐ = ί"'ΡΡ) ZVf (HZ <z
H 90"9=f ') 66·ε (H ε ε9·ε - sex (H ι 'ZH W =i 'p) isx (H e SOT - ιζ· 2 (H ε 's) (H ε 's) w s (H ε so (H ε Ζ6·ΐ (H ε 08·ΐ (Η ε 'ZH εε = f 'ρ) ει·ΐ (Η ε 'ΖΗ εε =1" 'ρ) ινι a g (Ρ- Ο ΟΗΟΊΗ 'ζ οοε) Η匪 HI H 90''9 = f ') 66 · ε (H ε ε9 · ε-sex (H ι' ZH W = i 'p) isx (H e SOT-ιζ · 2 (H ε' s) (H ε 's ) ws (H ε so (H ε Ζ6 · ΐ (H ε 08 · ΐ (Η ε 'ZH εε = f' ρ) ει · ΐ (Η ε ' Ζ εεε = 1''' ρ) ινι ag (Ρ- Ο ΟΗΟΊΗ ' ζ οοε) Η 匪 HI
°^ ¾(%εε Ό) ^m ^mM^ (I) 9P} ° ^ ¾ (% εε Ό) ^ m ^ mM ^ (I) 9P}
¾?第ェ、 ¾ί¾ 一ェ ^エ rci :- s - べ:^^
 / 一 α— ϋ / 一 ε— /— ½ 一 m— 一 g—[ べ:^¾? 第 、 第 ¾ί¾ 一 e ^ エ rci:-s-be: ^^ / One α— ϋ / One ε— / — ½ One m— One g— [B: ^
/ 一 α— ϋ / 一 ε— /— ½ 一 m— 一 g—[ べ:^¾? 第 、 第 ¾ί¾ 一 e ^ エ rci:-s-be: ^^ / One α— ϋ / One ε— / — ½ One m— One g— [B: ^
/^-ζ- ( ^ ェ {,^ [ ^エ( /;^ベ:^)—s] [ζπο / ^-ζ- (^ é {, ^ [^ e (/; ^ be: ^) — s] [ζπο

[9θ] [mo] έ / 一 α— ϋ / 一 ε— /— ½ 一 m— 一 g—[ / [9θ] [mo] έ / One α— ϋ / One ε— / — ½ One m— One g— [/
8圏第 [οπο]8th rank [οπο]
。 · コ 挲 ¾ 一^ S 、 ¾ 一 Ή Ν ¾ ο)ί¾?^ ^¾ °-Μ (% . · 挲 ¾ ¹ ^ S, ¾ Ή ¾ ¾ ο) ί¾? ^ ^ ¾ ° -Μ (%
8 、 90
 、つ攝慰 (I:S:(H =氺: 一, ェ: 8, 90 , Consolation (I: S: (H = 氺: ichi, é:
、つ攝慰 (I:S:(H =氺: 一, ェ: 8, 90 , Consolation (I: S: (H = 氺: ichi, é:
98C6S0/.00Zdf/X3d εε 8996ΖΪ/.00Ζ OAV
ピラゾール一 3—ィル 2,3,4,6—テトラ一 O ァセチル一 5 チォ一 β— D—ダルコ ビラノシドの代わりに 4— [4— (2- {ビス [2— (ベンジルォキシ)ェチル]アミノ}ェトキ シ) 2—メチルベンジル]—5—イソプロピル一 1H ピラゾールー 3—ィル 5 チォ β D ダルコビラノシドを用いて実施例 2 (2)と同様の方法で表題ィ匕合物を得た (0. 052g、 94%) o得られた化合物の構造、 NMRデータ及び MSデータを表 1に示 す。 98C6S0 / .00Zdf / X3d εε 8996ΖΪ / .00Ζ OAV Pyrazole 1-yl 2,3,4,6-tetra-1 O acetyl 1 5 thio 1 β- D-Dalco instead of bilanoside 4— [4— (2- {bis [2- (benzyloxy) ethyl] amino } Etoxy) 2-Methylbenzyl] -5-isopropyl 1H pyrazole-3-yl 5 thio β D darcobilanoside was used in the same manner as in Example 2 (2) to give the title compound (0. (052g, 94%) o The structure, NMR data and MS data of the obtained compound are shown in Table 1.
実施例 9 Example 9
N- [2 ヒドロキシ 1—(ヒドロキシメチル) 1ーメチルェチル]—N, {2— [4 ( 一イソプロピル一 3— [ (5—チォ一 13— D—ダルコピラノシル)ォキシ ]— 1H ピ ラゾール— 4—ィル }メチル)—3—メチルフヱノキシ]ェチル }尿素の製造 N- [2 Hydroxy 1- (hydroxymethyl) 1-methylethyl] -N, {2— [4 (Isopropyl-1-3- ((5-Thi-13-D-Darcopyranosyl) oxy]) — 1H Pyrazole 4-— R} methyl) -3-methylphenoxy] ethyl} Urea production
[0113] [化 17] [0113] [Chemical 17]

[0114] 2 アミノー 2—メチル 1—プロパノールの代わりに 2 アミノー 2—メチル 1,3— プロパンジオールを用い、実施例 7と同様の方法で表題ィ匕合物を得た (0. 123g、 86 %)。得られたィ匕合物の構造、 NMRデータ及び MSデータを表 1に示す。 [0114] The title compound was obtained in the same manner as in Example 7 except that 2-amino-2-methyl-1,3-propanediol was used instead of 2-amino-2-methyl-1-propanol (0.123 g, 86 %). Table 1 shows the structure, NMR data and MS data of the obtained compound.
実施例 10 Example 10
4- (4- {2- [ (2-ヒドロキシ - 1,1 -ジメチルェチル)ァミノ]エトキシ } 2 メチ ルベンジル) 5—イソプロピル一 1H ピラゾールー 3—ィル 5 チォ一 13 -D- ダルコビラノシドの製造 4- (4- {2- [(2-Hydroxy-1,1-dimethylethyl) amino] ethoxy} 2 methylbenzyl) 5-isopropyl-1- 1H pyrazole-3-yl 5-thio-13-D- Preparation of darcobilanoside
[0115] [化 18] [0115] [Chemical 18]

実施例 11 Example 11
N— [4— ( 一イソプロピル一 3— [ (5—チォ一 β—D—ダルコピラノシル)ォキシ] 1 Η ピラゾール 4ーィル }メチル)フエ-ル]尿素の製造 N— [4 -— (Isopropyl-1-3-((5-Thi-β-D-Darkopyranosyl) oxy] 1Ηpyrazole 4-methyl} methyl) phenol] urea
[0117] [化 19] [0117] [Chemical 19]

[0118] (1) 4— (4 ァミノベンジル) 5—イソプロピル一 1H ピラゾール一 3—ィル 2,3,4 6—テトラー Ο ァセチルー 5—チォー β D—ダルコビラノシドの合成
2, 3, 4, 6—テトラ一 O ァセチルー 5 チォ一 D—ダルコビラノース(25. 2g、 69 . Ommol)、 4— [ (4 -トロフエ-ル)メチル ] 1,2 ジヒドロ一 5—イソプロピル一 3 H ピラゾールー 3 オン(9. 02g、 34. 5mmol;国際特許公開 WO2004/019958号 を参考に-トロベンジルブロミドより合成)及びトリフエ-ルホスフィン(18. lg、 69. 0 mmol)のテトラヒドロフラン(43mL)溶液に氷冷下、ジイソプロピルァゾジカルボキシ レートの 40%トルエン溶液(36. 3mL、 69. Ommol)をゆっくりと滴下した。氷冷下で 30分間攪拌し、室温に昇温後、さらに 3時間攪拌した。溶媒を減圧下留去し、得られ た残渣をシリカゲルカラムクロマトグラフィ一(n—へキサン:酢酸ェチル = 1: 1)で精 製した。さらに、得られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム:メタ ノール = 50 : 1)にて精製し、 11. 9gの残渣を得た。この残渣のメタノール(195mL) 溶液に 10%パラジウム 活性炭素(3. 9g)を加え、水素雰囲気下、室温にてー晚攪 拌した。反応液をセライト濾過後、溶媒を減圧下留去し、得られた残渣をシリカゲル力 ラムクロマトグラフィー(へキサン:酢酸ェチル = 1: 3)で精製した。得られた残渣を再 度シリカゲルクロマトグラフィー(クロ口ホルム:メタノール = 20 : 1)にて精製し、淡黄色 液体として表題化合物(2. 68g、 13%)を得た。 [0118] (1) 4- (4-Aminobenzyl) 5-Isopropyl-1-H 1-Pyrazole 3-yl 2,3,4 6-Tetra Ο Acetyl-5-thio-β D-Dalcobilanoside Synthesis 2, 3, 4, 6-tetra-O-acetyl-5-thio-D-darcoviranose (25.2 g, 69. Ommol), 4-[(4-trifluoro) methyl] 1,2 dihydro-5-isopropyl 1 3 H pyrazol-3-one (9.02 g, 34.5 mmol; synthesized from trobenzyl bromide with reference to WO 2004/019958) and triphenylphosphine (18. lg, 69.0 mmol) in tetrahydrofuran ( (43 mL) A 40% toluene solution of diisopropylazodicarboxylate (36.3 mL, 69. Ommol) was slowly added dropwise to the solution under ice-cooling. The mixture was stirred for 30 minutes under ice-cooling, warmed to room temperature, and further stirred for 3 hours. The solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography (n-hexane: ethyl acetate = 1: 1). Furthermore, the obtained residue was purified by silica gel column chromatography (black mouth form: methanol = 50: 1) to obtain 19.9 g of residue. To a solution of this residue in methanol (195 mL) was added 10% palladium activated carbon (3.9 g), and the mixture was stirred at room temperature in a hydrogen atmosphere. The reaction mixture was filtered through celite, the solvent was evaporated under reduced pressure, and the obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 1: 3). The obtained residue was purified again by silica gel chromatography (chloroform: methanol = 20: 1) to obtain the title compound (2.68 g, 13%) as a pale yellow liquid.
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.15 (d, J=1.24 Hz, 3 H) 1.18 (d, J =1.24 Hz, 3 H) 1.89 (s, 3 H) 1.99 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.84 - 2.98 (m , 1 H) 3.29 - 3.38 (m, 1 H) 3.44 - 3.60 (m, 2 H) 4.07 - 4.17 (m, 1 H) 4.34 (dd, J=ll .97, 4.66 Hz, 1 H) 5.09 - 5.19 (m, 1 H) 5.38 (dd, J=10.10, 9.33 Hz, 1 H) 5.54 (t, J= 8.70 Hz, 1 H) 5.82 (d, J=8.70 Hz, 1 H) 6.58 (d, J=8.55 Hz, 2 H) 6.90 (d, J=8.55 Hz, 2 H). 1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.15 (d, J = 1.24 Hz, 3 H) 1.18 (d, J = 1.24 Hz, 3 H) 1.89 (s, 3 H) 1.99 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.84-2.98 (m, 1 H) 3.29-3.38 (m, 1 H) 3.44-3.60 (m, 2 H) 4.07-4.17 (m, 1 H) 4.34 (dd, J = ll .97, 4.66 Hz, 1 H) 5.09-5.19 (m, 1 H) 5.38 (dd, J = 10.10, 9.33 Hz, 1 H) 5.54 (t, J = 8.70 Hz, 1 H ) 5.82 (d, J = 8.70 Hz, 1 H) 6.58 (d, J = 8.55 Hz, 2 H) 6.90 (d, J = 8.55 Hz, 2 H).
(2) N— [4—( 一イソプロピル一 3— [ (5 チォ一 13—D—ダルコピラノシル)ォキ シ]— 1H ピラゾール— 4 ィル }メチル)フエ-ル]尿素の合成 (2) Synthesis of N— [4-((Isopropyl) -1-((5-thio-13-D-darcopyranosyl) oxy]]-1H-pyrazole- 4-yl} methyl) phenol] urea
4— [4— (2 アミノエトキシ) 2—メチルベンジル]—5—イソプロピル一 1H—ビラ ゾール—3—ィル 2, 3, 4, 6—テトラ— O ァセチル— 5 チォ— 13— D—ダルコ ビラノシドの代わりに 4— (4 ァミノベンジル) 5—イソプロピノレー 1H ピラゾーノレ 3—ィノレ 2,3,4,6—テトラー O ァセチノレー 5 チォー β D—ダルコビラノシドを 、 2 アミノー 2—メチルー 1 プロパノールの代わりにアンモニア(7Μメタノール溶
液)を用いて実施例 7と同様の方法で表題ィ匕合物(0. 125g、 73%)を得た。得られ た化合物の構造、 NMRデータ及び MSデータを表 1に示す。 4— [4— (2 Aminoethoxy) 2-methylbenzyl] —5-Isopropyl 1H-virazol-3-yl 2, 3, 4, 6-tetra-O-acetyl- 5 thio—13— D-Dalco 4- (4-aminobenzyl) 5-isopropylinole 3H-inole 2,3,4,6-tetra-O acetyleno 5 thio β D-darcobilanoside instead of viranoside, ammonia instead of 2-amino-2-methyl-1 propanol 7% methanol soluble The title compound (0.125 g, 73%) was obtained in the same manner as in Example 7. The structure, NMR data and MS data of the obtained compound are shown in Table 1.
実施例 12 Example 12
N— [4— ( 一イソプロピル一 3— [ (5—チォ一 13—D—ダルコピラノシル)ォキシ] — 1H ピラゾール— 4—ィル }メチル)フエ-ル] N, - (ピリジン— 3—ィルメチル) 尿素の製造 N— [4— (Isopropyl-1-3 — [(5-Thi-13-D-Darcopyranosyl) oxy] — 1H Pyrazole—4-yl} methyl) phenol] N,-(Pyridine-3-ylmethyl) Urea production
[0119] [化 20] [0119] [Chemical 20]
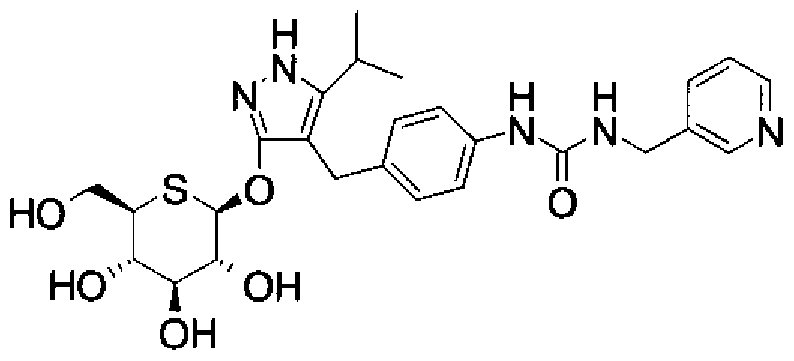
[0120] アンモニア(7Mメタノール溶液)の代わりに 3— (アミノメチル)ピリジンを用いて実施 例 11と同様の方法で表題ィ匕合物(0. 087g、 46%)を得た。得られた化合物の構造 、 NMRデータ及び MSデータを表 1に示す。 [0120] The title compound (0.087 g, 46%) was obtained in the same manner as in Example 11 except that 3- (aminomethyl) pyridine was used instead of ammonia (7M methanol solution). The structure, NMR data and MS data of the obtained compound are shown in Table 1.
実施例 13 Example 13
(3E) N— (2 ヒドロキシ— 1, 1 ジメチルェチル)—4— [4— ( —イソプロピル —3— [ (5 チォ一 13—D—ダルコピラノシル)ォキシ ] 1Η ピラゾール一 4—ィル }メチル)フエ-ル]ブタ— 3—ェンアミドの製造 (3E) N— (2 Hydroxy-1, 1 dimethylethyl) —4— [4— (—Isopropyl —3— [(5 thio 13-D-darcopyranosyl) oxy] 1Η pyrazole 1 -4-yl} methyl) -Lu] Buta-3-enamide production
[0121] [化 21] [0121] [Chemical 21]

(1) 4— (4 ブロモベンジル) 5—イソプロピノレ一 1H ピラゾーノレ一 3—ィノレ 5— チォー β D—ダルコビラノシドの合成 (1) 4- (4 Bromobenzyl) 5-Isopropinoleol 1H Pyrazolenol 3-Inole 5—Chiol β D—Dalcobilanoside
4 [ (4 -トロフエ-ル)メチル] 1,2 ジヒドロ 5 イソプロピル 3Η ピラゾ ール 3 オンの代わりに 4— [ (4 ブロモフエ-ル)メチル] 1,2 ジヒドロ 5 ィ ソプロピル 3Η ピラゾールー 3 オン(国際特許公開 WO2004/014932号を参考
に 4 ブロモベンジルブロミドより合成)を用い、実施例 11 (1)と同様の方法で 23. 4 gの残渣を得た。 4 [(4-Trophenyl) methyl] 1,2 dihydro-5 isopropyl 3Η instead of pyrazol 3 on 4— [(4 bromophenol) methyl] 1,2 dihydro 5 isopropyl 3Η pyrazol 3 on (international Patent publication Refer to WO2004 / 014932 23.4 g of a residue was obtained in the same manner as in Example 11 (1).
[0123] この残渣をメタノール(170mL)に溶解させ、ナトリウムメトキシド(4. 6Mメタノール 溶液、 15mL、 68mmol)を加え、 3時間攪拌した。反応液を濃縮後、残渣をシリカゲ ルクロマトグラフィー(クロ口ホルム:メタノール = 17 : 3)にて精製し、表題化合物(6. 7 4gゝ 27%)を得た。 [0123] This residue was dissolved in methanol (170 mL), sodium methoxide (4.6 M methanol solution, 15 mL, 68 mmol) was added, and the mixture was stirred for 3 hr. After the reaction solution was concentrated, the residue was purified by silica gel chromatography (black mouth form: methanol = 17: 3) to obtain the title compound (6.74 g 27%).
[0124] 1H NMR (600 MHz, METHANOL— d4) δ ppm 1.14 (dd, J=7.11, 3.90 Hz, 6 H) 2.8 2 - 2.93 (m, 2 H) 3.26 (t, J=8.94 Hz, 1 H) 3.52 - 3.68 (m, 2 H) 3.68 - 3.81 (m, 3 H ) 3.90 (dd, J=11.46, 3.67 Hz, 1 H) 5.45 (d, J=8.71 Hz, 1 H) 7.11 (d, J=8.25 Hz, 2 H ) 7.36 (d, J=8.25 Hz, 2 H). [0124] 1H NMR (600 MHz, METHANOL— d4) δ ppm 1.14 (dd, J = 7.11, 3.90 Hz, 6 H) 2.8 2-2.93 (m, 2 H) 3.26 (t, J = 8.94 Hz, 1 H ) 3.52-3.68 (m, 2 H) 3.68-3.81 (m, 3 H) 3.90 (dd, J = 11.46, 3.67 Hz, 1 H) 5.45 (d, J = 8.71 Hz, 1 H) 7.11 (d, J = 8.25 Hz, 2 H) 7.36 (d, J = 8.25 Hz, 2 H).
(2) 4- (4 ブロモベンジル) 5—イソプロピノレ一 1H ピラゾーノレ一 3—ィノレ 2,3, 4,6—テトラー O ァセチルー 5—チォー 13 D ダルコビラノシドの合成 (2) 4- (4 Bromobenzyl) 5-Isopropinoleol 1H Pyrazolenol 3-Inole 2,3,4,6-Tetra-O-acetylyl 5-Tio 13 D Synthesis of darcobilanoside
4— (4 ブロモベンジル)—5—イソプロピル— 1H ピラゾールー 3—ィル 5 チ ォ一 13—D—ダルコビラノシド(6. 25g、 13. 2mmol)のピリジン(50mL)溶液に無 水酢酸(25mL)をカ卩え、 2時間攪拌した。反応液に水を加え、酢酸ェチルで 2回抽 出後、有機層を 1N塩酸、飽和重曹水溶液、飽和食塩水で洗浄し、硫酸マグネシゥ ムで乾燥した。乾燥剤をろ別後、溶媒を減圧下留去した。残渣をシリカゲルクロマトグ ラフィー(へキサン:酢酸ェチル = 3 : 2)にて精製し、表題化合物(6. 08g、 72%)を 得た。 4- (4 bromobenzyl) -5-isopropyl-1H pyrazole-3-yl 5 thio 13-D-darcoviranoside (6.25 g, 13.2 mmol) in pyridine (50 mL) with anhydrous acetic acid (25 mL) Stir and stir for 2 hours. Water was added to the reaction mixture, and the mixture was extracted twice with ethyl acetate. The organic layer was washed with 1N hydrochloric acid, saturated aqueous sodium hydrogen carbonate solution and saturated brine, and dried over magnesium sulfate. After the desiccant was filtered off, the solvent was distilled off under reduced pressure. The residue was purified by silica gel chromatography (hexane: ethyl acetate = 3: 2) to obtain the title compound (6.08 g, 72%).
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.15 - 1.22 (m, 6 H) 1.87 (s, 3 H) 1.99 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.84 - 2.99 (m, 1 H) 3.32 - 3.44 (m, 1 H) 3 .50 - 3.66 (m, 2 H) 4.05 - 4.20 (m, 1 H) 4.34 (dd, J=11.97, 4.82 Hz, 1 H) 5.07 - 5. 22 (m, 9.09 Hz, 1 H) 5.32 - 5.45 (m, 1 H) 5.52 (t, J=8.70 Hz, 1 H) 5.85 (d, J=8.70 Hz, 1 H) 7.00 (d, J=8.39 Hz, 2 H) 7.35 (d, J=8.39 Hz, 2 H). 1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.15-1.22 (m, 6 H) 1.87 (s, 3 H) 1.99 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.84 -2.99 (m, 1 H) 3.32-3.44 (m, 1 H) 3.50-3.66 (m, 2 H) 4.05-4.20 (m, 1 H) 4.34 (dd, J = 11.97, 4.82 Hz, 1 H ) 5.07-5. 22 (m, 9.09 Hz, 1 H) 5.32-5.45 (m, 1 H) 5.52 (t, J = 8.70 Hz, 1 H) 5.85 (d, J = 8.70 Hz, 1 H) 7.00 ( d, J = 8.39 Hz, 2 H) 7.35 (d, J = 8.39 Hz, 2 H).
(3) (3E)—4— [4— ( 一イソプロピル一 3— [ (2,3,4,6—テトラ一 O ァセチル一 5 —チォ— 13—D—ダルコピラノシル)ォキシ]— 1H ピラゾール— 4—ィル }メチル) フエ-ル]ブター 3—ェン酸の合成 (3) (3E) —4— [4— (Isopropyl-1-3- (2,3,4,6-Tetra-O-acetyl-5-thio-13-D-darcopyranosyl) oxy] — 1H pyrazole— 4 —Yl} Methyl) Phenol] Butter 3-Synthesis
4— (4 ブロモベンジル) 5—イソプロピル一 1H ピラゾールー 3—ィル 2,3,4,
6—テトラ一 O ァセチルー 5 チォ一 β—D—ダルコビラノシド(0. 66g、 1. 03mm ol)のァセトニトリル(10mL)溶液にビニル酢酸(0. 21mL、 2. 47mmol)、酢酸パラ ジゥム(II) (22mg、 0. 10mmol)、トリ一 O トリノレホスフィン(60mg、 0. 20mmol)、 トリエチノレアミン(0. 72mL、 5. 15mmol)をカ卩え、 biotage社製マイクロウエーブを用 いて 120°C、 20分間反応を行った。反応液を減圧下留去し、得られた残渣をシリカ ゲルカラムクロマトグラフィー(クロ口ホルム:メタノール = 20 : 1)にて精製した。得られ た残渣を再度シリカゲルクロマトグラフィー(へキサン:酢酸ェチル = 1 :4)にて精製し 、淡黄色粉末として表題ィ匕合物(301mg、 56%)を得た。 4- (4 Bromobenzyl) 5-Isopropyl 1H Pyrazole-3-yl 2,3,4, 6-tetra-O-acetyl-5-thio-β-D-darcoviranoside (0.66 g, 1.03 mmol) in acetonitrile (10 mL) with vinylacetic acid (0.21 mL, 2.47 mmol), paradium acetate (II) ( 22 mg, 0.10 mmol), tri-O-trinolephosphine (60 mg, 0.20 mmol), tritinoreamine (0.72 mL, 5.15 mmol), 120 ° C using biotage microwave The reaction was performed for 20 minutes. The reaction solution was evaporated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (black mouth form: methanol = 20: 1). The obtained residue was purified again by silica gel chromatography (hexane: ethyl acetate = 1: 4) to obtain the title compound (301 mg, 56%) as a pale yellow powder.
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.13 - 1.20 (m, J=6.99, 2.95 Hz, 6 H) 1.85 (s, 3 H) 1.99 (s, 3 H) 2.03 (s, 3 H) 2.05 (s, 3 H) 2.82 - 2.96 (m, 1 H) 3.21 - 3.37 (m, 3 H) 3.54 - 3.68 (m, J=3.89 Hz, 2 H) 4.09 - 4.18 (m, J=12.59, 3.73 Hz, 1 H) 4.33 (dd, J=11.73, 4.90 Hz, 1 H) 5.09 - 5.19 (m, 1 H) 5.31 - 5.42 (m, 1 H) 5.5 3 (t, J=8.55 Hz, 1 H) 5.73 (d, J=8.55 Hz, 1 H) 6.16 - 6.30 (m, 1 H) 6.41 - 6.55 (m, 1 H) 7.06 (d, J=7.77 Hz, 2 H) 7.22 - 7.28 (m, 2 H). 1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.13-1.20 (m, J = 6.99, 2.95 Hz, 6 H) 1.85 (s, 3 H) 1.99 (s, 3 H) 2.03 (s, 3 H) 2.05 (s, 3 H) 2.82-2.96 (m, 1 H) 3.21-3.37 (m, 3 H) 3.54-3.68 (m, J = 3.89 Hz, 2 H) 4.09-4.18 (m, J = 12.59, 3.73 Hz , 1 H) 4.33 (dd, J = 11.73, 4.90 Hz, 1 H) 5.09-5.19 (m, 1 H) 5.31-5.42 (m, 1 H) 5.5 3 (t, J = 8.55 Hz, 1 H) 5.73 (d, J = 8.55 Hz, 1 H) 6.16-6.30 (m, 1 H) 6.41-6.55 (m, 1 H) 7.06 (d, J = 7.77 Hz, 2 H) 7.22-7.28 (m, 2 H) .
(4) (3E) -N- (2 ヒドロキシ一 1, 1 ジメチルェチル) 4— [4— ( 一イソプロピ ル一 3— [ (2,3,4,6—テトラ一 O ァセチル一 5—チォ一 13—D—ダルコピラノシル) ォキシ] 1H ピラゾール— 4 ィル }メチル)フエ-ル]ブタ— 3 ェンアミドの合成 (3E)—4— [4— ( 一イソプロピル一 3— [ (2,3,4,6—テトラ一 O ァセチル一 5— チォ一 13—D—ダルコピラノシル)ォキシ]— 1H ピラゾール一 4—ィル }メチル)フエ -ル]ブタ— 3 ェン酸(0. 144g、0. 223mmol)の N, N ジメチルホルムアミド(1 . 5mL)溶液に、 2 アミノー 2—メチノレ一 1—プロノノーノレ(31. 9 /z L、0. 334mmo 1)、 1—ヒドロキシベンゾトリアゾール 1水和物(90mg、 0. 668mmol)、 N ェチルー N' — 3 ジメチルァミノプロピルカルボジイミド塩酸塩( 128mg、 0. 668mmol)を 加え、室温にて 2時間攪拌した。反応液をシリカゲルカラムクロマトグラフィ一(クロロホ ルム:メタノール = 20 : 1)にて精製し、淡黄色粉末として表題化合物(138mg、 88% )を得た。 (4) (3E) -N- (2 Hydroxy 1,1, 1 dimethylethyl) 4— [4— (Monoisopropyl 3- 3- [(2,3,4,6-Tetra-O-acetyl 1-5-thio 13) —D—Darcopyranosyl) oxy] 1H pyrazole—4yl} methyl) phenol] buta-3-enamide synthesis (3E) —4— [4— (Isopropyl-3-[(2,3,4,6 —Tetra-1-O-acetyl-l 5—thio-l 13-D-darcopyranosyl) oxy] —— 1H pyrazole-l-yl} methyl) phenol] buta-3-enoic acid (0.144 g, 0.223 mmol) N , N in dimethylformamide (1.5 mL), 2-amino-2-methylolone 1-prononanol (31.9 / z L, 0.334 mmo 1), 1-hydroxybenzotriazole monohydrate (90 mg, 0.668 mmol) ), Nethyl-N′—3 dimethylaminopropylcarbodiimide hydrochloride (128 mg, 0.668 mmol) was added, and the mixture was stirred at room temperature for 2 hours. The reaction solution was purified by silica gel column chromatography (chloroform: methanol = 20: 1) to obtain the title compound (138 mg, 88%) as a pale yellow powder.
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.18 (d, J=7.15 Hz, 6 H) 1.28 (s, 6 H) 1.87 (s, 3 H) 1.99 (s, 3 H) 2.03 (s, 3 H) 2.05 (s, 3 H) 3.06 - 3.16 (m, 2 H) 3.3
2 - 3.44 (m, 1 H) 3.53 - 3.70 (m, 4 H) 4.05 - 4.18 (m, 2 H) 4.33 (dd, J=11.97, 4.82 Hz, 1 H) 5.11 - 5.22 (m, 1 H) 5.32 - 5.42 (m, 1 H) 5.53 (t, J=8.70 Hz, 1 H) 5.86 (d,1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.18 (d, J = 7.15 Hz, 6 H) 1.28 (s, 6 H) 1.87 (s, 3 H) 1.99 (s, 3 H) 2.03 (s, 3 H) 2.05 (s, 3 H) 3.06-3.16 (m, 2 H) 3.3 2-3.44 (m, 1 H) 3.53-3.70 (m, 4 H) 4.05-4.18 (m, 2 H) 4.33 (dd, J = 11.97, 4.82 Hz, 1 H) 5.11-5.22 (m, 1 H) 5.32-5.42 (m, 1 H) 5.53 (t, J = 8.70 Hz, 1 H) 5.86 (d,
J=8.70 Hz, 1 H) 6.11 - 6.26 (m, 1 H) 6.48 (d, J=15.85 Hz, 1 H) 7.08 (d, J=8.24 Hz,J = 8.70 Hz, 1 H) 6.11-6.26 (m, 1 H) 6.48 (d, J = 15.85 Hz, 1 H) 7.08 (d, J = 8.24 Hz,
2 H) 7.25 (d, J=8.24 Hz, 2 H). 2 H) 7.25 (d, J = 8.24 Hz, 2 H).
(5) (3E) -N- (2 ヒドロキシ一 1, 1 ジメチルェチル) 4— [4— ( 一イソプロピ ル一 3— [ (5—チォ一 13—D—ダルコピラノシル)ォキシ ] 1Η ピラゾール一 4—ィ ル}メチル)フ -ル]ブター 3—ェンアミドの合成 (5) (3E) -N- (2 Hydroxy 1,1, dimethylethyl) 4— [4— (Monoisopropyl 3- 3- [(5-Thi 13-D-Dalcopyranosyl) oxy] 1Η Pyrazole 4- 4- Synthesis of Ru} methyl) fur] buter 3-enamide
(3E)— N— (2—ヒドロキシ— 1, 1 ジメチルェチル)—4— [4— ( —イソプロピル —3— [ (2,3,4,6—テトラ一 O ァセチル一 5—チォ一 13—D—ダルコピラノシル)ォ キシ] 1H ピラゾール— 4 ィル }メチル)フエ-ル]ブタ— 3 ェンアミド(0. 124g 、 0. 176mmol)のメタノール(2mL)溶液に、ナトリウムメトキシド(25%メタノール溶 液; 0. 38mL、 0. 18mmol)を加え、室温で 2時間攪拌した。反応液を減圧濃縮後、 残渣をシリカゲルカラムクロマトグラフィ (酢酸ェチル:エタノール:水 = 20: 2: 1)で 精製し、表題ィ匕合物(63mg、 65%)を合成した。得られた化合物の構造、 NMRデ ータ及び MSデータを表 1に示す。 (3E) — N— (2-Hydroxy— 1, 1 Dimethylethyl) —4— [4— (— Isopropyl —3— [(2,3,4,6—Tetra-I O-acetyl-5- 5-thio 13-D —Darkopyranosyl) oxy] 1H pyrazole—4yl} methyl) phenol] buta-3-enamide (0.124 g, 0.176 mmol) in methanol (2 mL) in sodium methoxide (25% methanol solution; (0.38 mL, 0.18 mmol) was added, and the mixture was stirred at room temperature for 2 hours. The reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate: ethanol: water = 20: 2: 1) to synthesize the title compound (63 mg, 65%). Table 1 shows the structure, NMR data and MS data of the compound obtained.
実施例 14 Example 14
(3E)— N— (2 アミノー 1, 1 ジメチルー 2—ォキソェチル) 4— [4— ( 一イソ プロピル一 3— [ (5—チォ一 13—D—ダルコピラノシル)ォキシ] 1H ピラゾール —4—ィル }メチル)フエ-ル]ブタ— 3—ェンアミドの製造 (3E) — N— (2 Amino-1, 1 Dimethyl-2-oxoethyl) 4— [4— (Monoisopropyl I 3— [(5-Dio 13-D-Darkopyranosyl) oxy] 1H Pyrazole —4-yl } Methyl) phenol] but-3-enamide production
[0126] [化 22] [0126] [Chemical 22]

[0127] (1) (3E)— N— (2 アミノー 1, 1 ジメチルー 2—ォキソェチル) 4— [4 5—ィ ソプロピル一 3— [ (2,3,4,6—テトラ一 O ァセチルー 5—チォ一 13—D—ダルコビラ ノシル)ォキシ]― 1H ピラゾール— 4—ィル }メチル)フエ-ル]ブタ— 3 ェンアミド の合成 [0127] (1) (3E) — N— (2 Amino-1, 1 Dimethyl-2-oxoethyl) 4— [4 5—Sopropyl 1 3— [(2,3,4,6—Tetra 1 O acetylyl 5— 13-D-Dalcoviranosyl) oxy] -1H pyrazole-4-yl} methyl) phenol] buta-3-enamide
2—アミノー 2—メチル 1—プロパノールの代わりに 2—メチルァラニンアミドを用い
て、実施例 13 (4)と同様の方法で表題ィ匕合物(0. 120g、 70%)を得た。 2-Methylalananamide instead of 2-amino-2-methyl 1-propanol In the same manner as in Example 13 (4), the title compound (0.120 g, 70%) was obtained.
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.15 (d, J=6.68 Hz, 6 H) 1.58 (d, J=2.80 Hz, 6 H) 1.85 (s, 3 H) 2.00 (s, 3 H) 2.03 (s, 1 H) 2.05 (s, 3 H) 2.79 - 3.03 ( m, 2 H) 3.12 (d, J=7.15 Hz, 2 H) 3.36 - 3.71 (m, 3 H) 4.05 - 4.19 (m, 2 H) 4.33 (dd , J=11.97, 4.66 Hz, 1 H) 5.13 - 5.27 (m, 1 H) 5.32 - 5.44 (m, 1 H) 5.49 - 5.60 (m, 1 H) 5.86 (d, J=8.70 Hz, 1 H) 6.13 - 6.29 (m, 1 H) 6.46 (d, J=15.85 Hz, 1 H) 7.06 ( d, J=7.93 Hz, 2 H) 7.23 (d, J=7.93 Hz, 2 H). 1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.15 (d, J = 6.68 Hz, 6 H) 1.58 (d, J = 2.80 Hz, 6 H) 1.85 (s, 3 H) 2.00 (s, 3 H) 2.03 (s, 1 H) 2.05 (s, 3 H) 2.79-3.03 (m, 2 H) 3.12 (d, J = 7.15 Hz, 2 H) 3.36-3.71 (m, 3 H) 4.05-4.19 (m, 2 H) 4.33 (dd, J = 11.97, 4.66 Hz, 1 H) 5.13-5.27 (m, 1 H) 5.32-5.44 (m, 1 H) 5.49-5.60 (m, 1 H) 5.86 (d, J = 8.70 Hz, 1 H) 6.13-6.29 (m, 1 H) 6.46 (d, J = 15.85 Hz, 1 H) 7.06 (d, J = 7.93 Hz, 2 H) 7.23 (d, J = 7.93 Hz, 2 H ).
(2) (3E) N— (2 アミノー 1, 1 ジメチルー 2—ォキソェチル)ー4 [4 ( ーィ ソプロピル一 3— [ (5 チォ一 13—D—ダルコピラノシル)ォキシ] 1H ピラゾール 4ーィル }メチル)フ -ル]ブター 3—ェンアミドの合成 (2) (3E) N— (2 amino-1, 1 dimethyl-2-oxoethyl) -4 [4 (-isopropyl-1-3- ((5-thiol-13-D-darcopyranosyl) oxy] 1H pyrazole 4-yl} methyl) Synthesis of [Fur] buter 3-enamide
(3E) N— (2 ヒドロキシ一 1, 1 ジメチルェチル) 4— [4— ( 一イソプロピル —3— [ (2,3,4,6—テトラ一 O ァセチル一 5—チォ一 13—D—ダルコピラノシル)ォ キシ] 1H ピラゾール 4 ィル }メチル)フエ-ル]ブタ一 3 ェンアミドの代わり に(3E)— N— (2 アミノー 1,1 ジメチルー 2—ォキソェチル) 4— [4 ( 一イソ プロピル一 3— [ (2,3,4,6—テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノ シル)ォキシ] - 1H ピラゾール— 4—ィル }メチル)フエ-ル]ブタ— 3 ェンアミドを 用いて、実施例 13 (5)と同様の方法で表題ィ匕合物(0. 067g、 73%)を得た。得られ た化合物の構造、 NMRデータ及び MSデータを表 1に示す。 (3E) N— (2 hydroxy-1,1,1 dimethylethyl) 4— [4— (monoisopropyl —3— [(2,3,4,6-tetra-1, O acetyl, 5-thio-1, 13-D-darcopyranosyl) Oxy] 1H pyrazole 4 methyl} methyl) phenol] but instead of 3 amide (3E) —N— (2 amino-1,1 dimethyl-2-oxohexyl) 4— [4 (monoisopropyl 3- [(2,3,4,6-tetra-O-acetyl-5-thio 13-D-darcopyranosyl) oxy] -1H pyrazole-4-yl} methyl) phenol] buta-3-enamide The title compound (0.067 g, 73%) was obtained in the same manner as in Example 13 (5). The structure, NMR data and MS data of the obtained compound are shown in Table 1.
実施例 15 Example 15
N— (2—ヒドロキシ一 1, 1 ジメチルェチル) 4— [4— ( 一イソプロピル一 3— [( 5 チォ— 13—D—ダルコピラノシル)ォキシ]— 1H ピラゾールー 4—ィル }メチル) フエ-ル]ブタンアミドの製造 N— (2-hydroxy-1,1,1 dimethylethyl) 4— [4— (monoisopropyl 1-3 — [(5 thio-13-D-darcopyranosyl) oxy] — 1H pyrazol-4-yl} methyl) phenol] Production of butanamide
[0129] [化 23] [0129] [Chemical 23]

[0130] (1) 4— [4— (15—イソプロピル一 3— [ (2,3,4,6—テトラ一 O ァセチル一 5 チォ
- β—D—ダルコピラノシル)ォキシ ] 1Η ピラゾール— 4—ィル }メチル)フエ-ル ]ブタン酸の合成 [0130] (1) 4— [4— (15—Isopropyl 1 3 — [(2,3,4,6—Tetra 1 O acetyl 1 5 thio -β-D-Darcopyranosyl) oxy] 1Ηpyrazole-4-yl} methyl) phenol] butanoic acid synthesis
実施例 13 (3)で合成した(3Ε)— 4— [4— ( 一イソプロピル一 3— [ (2,3,4,6—テ トラ一 Ο ァセチノレ一 5 チォ一 13—D—ダルコピラノシル)ォキシ ] 1Η ピラゾー ルー 4—ィル }メチル)フエ-ル]ブタ一 3 ェン酸(0. 728g、 1. 13mmol)のメタノー ル(5mL)溶液に 10%パラジウム—活性炭(70mg)を加え、水素雰囲気下でー晚攪 拌した。反応液をセライト濾過後、ろ液を濃縮し、表題化合物(0. 670g、 91%)を得 た。 Example 13 (3Ε) — 4— [4— (Isopropyl 1- 3 — [(2,3,4,6—Tetra 1 ァ Acetinore 1 5 Thio 13-D-Darcopyranosyl) oxy) ] 1Η Pyrazol® 4-yl} methyl) phenol] buta-3-enic acid (0.728 g, 1.13 mmol) in methanol (5 mL) was added 10% palladium-activated carbon (70 mg) and hydrogen Stirred in the atmosphere. The reaction mixture was filtered through celite, and the filtrate was concentrated to give the title compound (0.670 g, 91%).
[0131] 1H NMR (300 MHz, CHLOROFORM- d) δ ppm 1.15 (d, J=2.33 Hz, 3 H) 1.18 (d, [0131] 1H NMR (300 MHz, CHLOROFORM- d) δ ppm 1.15 (d, J = 2.33 Hz, 3 H) 1.18 (d,
J=2.33 Hz, 3 H) 1.84 - 1.95 (m, 2 H) 1.86 (s, 3 H) 1.99 (s, 3 H) 2.01 - 2.02 (m, 3 H) 2.04 (s, 3 H) 2.27 - 2.37 (m, 2 H) 2.54 - 2.67 (m, 2 H) 2.84 - 2.99 (m, 1 H) 3.2 7 - 3.38 (m, 1 H) 3.53 - 3.68 (m, 2 H) 4.13 (dd, J=11.97, 3.96 Hz, 1 H) 4.32 (dd, J =11.97, 4.82 Hz, 1 H) 5.09 - 5.19 (m, 1 H) 5.30 - 5.44 (m, 1 H) 5.53 (t, J=8.55 Hz, 1 H) 5.75 (d, J=8.55 Hz, 1 H) 7.03 (s, 4 H). J = 2.33 Hz, 3 H) 1.84-1.95 (m, 2 H) 1.86 (s, 3 H) 1.99 (s, 3 H) 2.01-2.02 (m, 3 H) 2.04 (s, 3 H) 2.27-2.37 (m, 2 H) 2.54-2.67 (m, 2 H) 2.84-2.99 (m, 1 H) 3.2 7-3.38 (m, 1 H) 3.53-3.68 (m, 2 H) 4.13 (dd, J = 11.97 , 3.96 Hz, 1 H) 4.32 (dd, J = 11.97, 4.82 Hz, 1 H) 5.09-5.19 (m, 1 H) 5.30-5.44 (m, 1 H) 5.53 (t, J = 8.55 Hz, 1 H ) 5.75 (d, J = 8.55 Hz, 1 H) 7.03 (s, 4 H).
(2) N- (2 ヒドロキシ一 1, 1 ジメチルェチル) 4— [4— ( 一イソプロピル一 3 — [(5 チォ一 13—D—ダルコピラノシル)ォキシ ] 1Η ピラゾール一 4—ィル }メチ ル)フ ニル]ブタンアミドの合成 (2) N- (2 hydroxy-1,1,1 dimethylethyl) 4- [4— (monoisopropyl 1-3 — [(5 thio 13-D-darcopyranosyl) oxy] 1Η pyrazole 4-yl} methyl) Of Nyl] butanamide
(3E)—4— [4— ( 一イソプロピル一 3— [ (2,3,4,6—テトラ一 O ァセチル一 5— チォ一 13—D—ダルコピラノシル)ォキシ]— 1H ピラゾール一 4—ィル }メチル)フエ -ル]ブタ一 3—ェン酸の代わりに 4— [4—( 一イソプロピル一 3— [ (2,3,4,6—テ トラ一 O ァセチノレ一 5 チォ一 13—D—ダルコピラノシル)ォキシ ] 1Η ピラゾー ル— 4 ィル }メチル)フエニル]ブタン酸を用いて、実施例 13 (4)と同様の方法で 0. 154gの残渣を得た。 (3E) —4— [4— (Isopropyl-1-3- ((2,3,4,6-Tetra-1-acetyl 5--thio 13-D-darcopyranosyl) oxy]] — 1H Pyrazole 4-yl } Methyl) phenol] buta-3-ethanoic acid instead of 4-[4- (monoisopropyl-1-3- ((2,3,4,6--tetra-O-acetinol-5-thio 13-D —Dalcopyranosyl) oxy] 1Ηpyrazole—4yl} methyl) phenyl] butanoic acid was used in the same manner as in Example 13 (4) to obtain 0.154 g of a residue.
[0132] この残渣のメタノール(2mL)溶液に、ナトリウムメトキシド(25%メタノール溶液; 52 [0132] To a solution of this residue in methanol (2 mL), sodium methoxide (25% methanol solution; 52
μレ 0. 24mmol)を加え、室温で 2時間攪拌した。反応液を減圧濃縮後、残渣をシ リカゲルカラムクロマトグラフィ (酢酸ェチル:エタノール:水 = 15: 2: 1)で精製し、 表題ィ匕合物(90mg、 68%)を合成した。得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。
実施例 16 μle 0.24 mmol) was added, and the mixture was stirred at room temperature for 2 hours. The reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate: ethanol: water = 15: 2: 1) to synthesize the title compound (90 mg, 68%). The structure, NMR data and MS data of the obtained compound are shown in Table 1. Example 16
N— (2 アミノー 1,1 ジメチノレ - 2-ォキソェチノレ) -4- [4- ({5-イソプロピル —3— [ (5 チォ一 13—D—ダルコピラノシル)ォキシ ] 1Η ピラゾール一 4—ィル }メチル)フエ-ル]ブタンアミドの製造 N— (2 Amino-1,1 Dimethinole-2-Oxochinole) -4- [4- ({5-Isopropyl —3— [(5 Thio 13-D-Dalcopyranosyl) oxy] 1Η Pyrazole 4-yl} methyl ) Fuel] butanamide production
[0133] [化 24] [0133] [Chemical 24]

[0134] 2 アミノー 2—メチルー 1 プロパノールの代わりに 2—メチルァラニンアミドを用い 、実施例 15 (2)と同様の方法で表題ィ匕合物(69mg、 54%)を得た。得られた化合物 の構造、 NMRデータ及び MSデータを表 1に示す。 [0134] The title compound (69 mg, 54%) was obtained in the same manner as in Example 15 (2) using 2-methylalaninamide instead of 2-amino-2-methyl-1-propanol. The structure, NMR data and MS data of the obtained compound are shown in Table 1.
実施例 17 Example 17
N- [2 ヒドロキシ 1, 1 ビス(ヒドロキシメチル)ェチル]ー4 [4 ( —イソプ 口ピル一 3— [(5—チォ一 13—D—ダルコピラノシル)ォキシ ] 1Η ピラゾールー 4 ーィル }メチル)フエ-ル]ブタンアミドの製造 N- [2 Hydroxy 1,1 bis (hydroxymethyl) ethyl] -4 [4 (—Isopropyl pill 1— [(5—Cho 13-D-Dalcopyranosyl) oxy] 1Η Pyrazole-4-yl} methyl) Lu] butanamide production
[0135] [化 25] [0135] [Chemical 25]
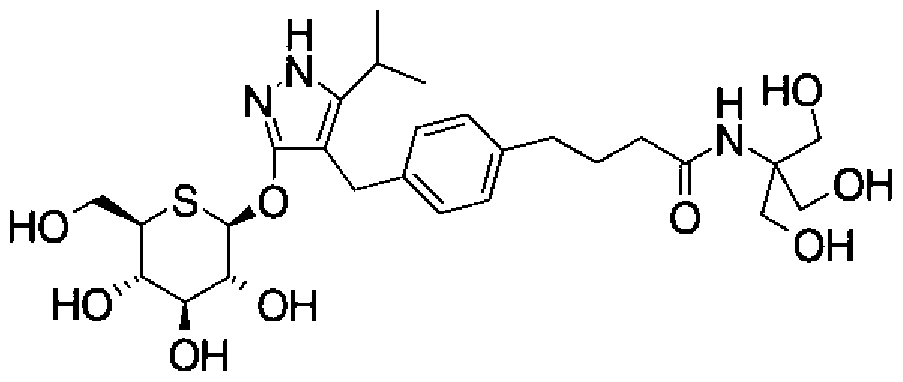
2 -ァミノ 2—メチル 1—プロパノールの代わりにトリス(ヒドロキシメチル)アミノメ タンを用い、実施例 15 (2)と同様の方法で表題ィ匕合物(9mg、 6%)を得た。得られ た化合物の構造、 NMRデータ及び MSデータを表 1に示す。 The title compound (9 mg, 6%) was obtained in the same manner as in Example 15 (2) using tris (hydroxymethyl) aminomethane in place of 2-amino-2-methyl-1-propanol. The structure, NMR data and MS data of the obtained compound are shown in Table 1.
実施例 18 Example 18
N— [1, 1 ジメチルー 2— (4—メチルビペラジン一 1—ィル) 2—ォキソェチル] —4— {4— ( 一イソプロピル一 3— [(5—チォ一 13—D—ダルコピラノシル)ォキシ] — 1 H ピラゾール 4 ィル }メチル)フエ-ル }ブタンアミドの製造
[0137] [化 26] N— [1, 1 Dimethyl-2- (4-methylbiperazine 1-yl) 2-oxoethyl] —4— {4— (Isopropyl-3- 3 [[(5-Thi-13-D-Darcopyranosyl) oxy] — 1 H Pyrazole 4-methyl) phenyl} butanamide [0137] [Chemical 26]

[0138] 2 アミノー 2—メチルー 1 プロパノールの代わりに 2—メチルー 1ー(4ーメチルビ ペラジン一 1—ィル )一 1 ォキソプロパン一 2 アミンを用い、実施例 15 (2)と同様の 方法で表題ィ匕合物(69mg、 54%)を得た。得られた化合物の構造、 NMRデータ及 び MSデータを表 1に示す。 [0138] Instead of 2-amino-2-methyl-1-propanol, 2-methyl-1- (4-methylbiperazine-1-yl) 1-1-oxopropane-2-amine was used in the same manner as in Example 15 (2). The compound (69 mg, 54%) was obtained. The structure, NMR data and MS data of the obtained compound are shown in Table 1.
[0139] [表 1-1]
[0139] [Table 1-1]
98£6S0.00v:zfcl>d S 8996Π O.00ΖAV 98 £ 6S0.00v: zfcl> d S 8996Π O.00ΖAV

Κ uΐ00-
/ 98£6so/-osa/:Tl£ 9899/ O62/-00ZAV Κ uΐ00- / 98 £ 6so / -osa /: Tl £ 9899 / O62 / -00ZAV



98f6£0/.00Zdf/X3d 817 8996ΖΪ/.00Ζ OAV
 98f6 £ 0 / .00Zdf / X3d 817 8996ΖΪ / .00Ζ OAV
98f6 £ 0 / .00Zdf / X3d 817 8996ΖΪ / .00Ζ OAV
[0144] 製剤実施例 [0145] [表 2]
薬物含量 lOOmg錠剤の処方: [0144] Formulation Examples [0145] [Table 2] Drug content lOOmg tablet formulation:
1錠中含量: Content in 1 tablet:
薬物 108.35 mg Drug 108.35 mg
乳糖一水和物 38.65 mg Lactose monohydrate 38.65 mg
結晶セル口一ス 22.00 mg Crystal cell mouthpiece 22.00 mg
カルボキシメチルセル口一スカル 20.00 mg Carboxymethyl cell mouth skull 20.00 mg
ヒド□キシプロピルセルロース 10.00 mg Hydoxypropyl cellulose 10.00 mg
ステアリン酸マグネシウム Magnesium stearate
[0146] 製造方法 [0146] Manufacturing method
薬物 (本発明化合物)を乳糖一水和物、結晶セルロース、カルボキシメチルセル口 —スカルシウム及びヒドロキシプロピルセルロースと混合し、この混合物を粉砕機で粉 砕する。粉砕された混合物を撹拌造粒機で 1分間混合し、その後、水で 4〜8分間造 粒する。得られた造粒物を 70°C、 40分間乾燥する。造粒乾燥末を 500 mの篩で 篩過する。篩過後の造粒乾燥末とステアリン酸マグネシウムを、 V型混合機を用いて 30rpmで 3分間混合する。口—タリ—式打錠機を用いて得られた打錠用顆粒を圧縮 成形し製錠する。 The drug (the compound of the present invention) is mixed with lactose monohydrate, crystalline cellulose, carboxymethyl cellulose-calcium and hydroxypropylcellulose, and this mixture is pulverized with a pulverizer. The pulverized mixture is mixed with a stirring granulator for 1 minute, and then granulated with water for 4-8 minutes. The resulting granulate is dried at 70 ° C for 40 minutes. Sift the granulated dry powder with a 500 m sieve. The granulated dry powder after sieving and magnesium stearate are mixed at 30 rpm for 3 minutes using a V-type mixer. The granules for tableting obtained using a mouth-tally tablet press are compression-molded and tableted.
[0147] [表 3] 錠剤重量: 200mg [0147] [Table 3] Tablet weight: 200mg
錠径 —: 8 mm, 円彤 Tablet diameter —: 8 mm, round bowl
[0148] 試験例 1 [0148] Test Example 1
(1)ヒト SGLT1とヒト SGLT2のクローユングと発現ベクターへの導入 (1) Cloning of human SGLT1 and human SGLT2 and introduction into expression vectors
ヒト小腸由来 mRNAからヒト SGLT1配列(NM— 000343)を逆転写の後増幅し、 pCMV— tag5A (ストラタジーン社)に導入した。また、ヒト SGLT2配列(NM— 003 041)はヒト腎由来 mRNAから同様な方法で調製し、 pcDNA3. 1 +hygro (インビト ロジェン社)に導入した。それぞれのクローンの配列力 報告されている配列と一致 することを確認した。 A human SGLT1 sequence (NM-000343) was amplified from human small intestine-derived mRNA after reverse transcription and introduced into pCMV-tag5A (Stratagene). A human SGLT2 sequence (NM-003 041) was prepared from human kidney-derived mRNA by the same method and introduced into pcDNA3.1 + hygro (Invitrogen). The sequence ability of each clone was confirmed to match the reported sequence.
(2)ヒト SGLT1及びヒト SGLT2を安定に発現する CHO— kl細胞の作成 ヒト SGLT1およびヒト SGLT2発現ベクターを、リポフエクトァミン 2000 (インビトロジ ェン社)を用いて CHO—K1細胞へトランスフエクシヨンした。 SGLT発現細胞は、 50
0 μ gZmLの濃度のジエネティシン(SGLT1)またはハイグロマイシン B (SGLT2)の 存在下で培養し耐性株を選択し、下記に示す系により糖取り込み比活性を指標に取 得した。 (2) Preparation of CHO-kl cells that stably express human SGLT1 and human SGLT2 Transfect human SGLT1 and human SGLT2 expression vectors into CHO-K1 cells using Lipofectamine 2000 (Invitrogen). did. SGLT expressing cells are 50 Resistant strains were selected by culturing in the presence of dieneticin (SGLT1) or hygromycin B (SGLT2) at a concentration of 0 μgZmL, and the glucose uptake specific activity was obtained using the system shown below as an index.
(3)細胞におけるナトリウム依存的糖取り込み阻害試験 (3) Sodium-dependent inhibition of glucose uptake in cells
ヒト SGLT1又はヒト SGLT2を安定に発現する細胞をナトリウム依存的グルコース取 り込み活性阻害試験に用いた。 Cells stably expressing human SGLT1 or human SGLT2 were used in a sodium-dependent glucose uptake activity inhibition test.
[0149] 前処理用緩衝液(140mM 塩化コリン、 2mM KC1、 ImM CaCl、 ImM Mg [0149] Pretreatment buffer (140 mM choline chloride, 2 mM KC1, ImM CaCl, ImM Mg
2 2
Cl、 10mM HEPES/5mM Tris、 pH7. 4) 200 Lをヒト SGLT1発現細胞に Cl, 10 mM HEPES / 5 mM Tris, pH 7.4) 200 L to human SGLT1-expressing cells
2 2
又は 2mLをヒト SGLT2発現細胞にカ卩え、 20分間インキュベーションした。前処理用 緩衝液を除去し、試験化合物を含む取り込み用緩衝液 ( ["C]メチル a— D—ダル コピラノシドを含むメチル α— D—ダルコビラノシド(lmM)、 145mM NaCl、 2m M KC1、 ImM CaCl、 ImM MgCl、 lOmM HEPES/5mM Tris、 pH7. 4 Alternatively, 2 mL was added to human SGLT2-expressing cells and incubated for 20 minutes. Pretreatment buffer is removed and uptake buffer containing test compound (["C] methyl α-D-dalcopyranoside containing α-D-darcobilanoside (lmM), 145 mM NaCl, 2 mM KC1, ImM CaCl , ImM MgCl, lOmM HEPES / 5mM Tris, pH7.4
2 2 twenty two
)を 75 L (SGLT1の場合)又は 200 μ L (SGLT2の場合)加え、 37°Cにて 30分 (S GLT1の場合)又は 1時間(SGLT2の場合)取り込み反応を行った。反応後細胞を 洗浄用緩衝液(lOmM メチル α— D—ダルコビラノシド、 140mM 塩化コリン 2m M KC1、 ImM CaCl、 ImM MgCl、 lOmM HEPES/5mM Tris、 pH7. 4 ) Was added at 75 L (in the case of SGLT1) or 200 μL (in the case of SGLT2), and an uptake reaction was performed at 37 ° C for 30 minutes (in the case of S GLT1) or 1 hour (in the case of SGLT2). After the reaction, the cells were washed with a buffer for washing (lOmM methyl α-D-darcobilanoside, 140 mM choline chloride 2 mM KCl, ImM CaCl, ImM MgCl, lOmM HEPES / 5 mM Tris, pH 7.4
2 2 twenty two
) 200 し(301/11の場合)又は2111し(301^2の場合)で2回洗浄し、0. 2M NaO H溶液 75 μ L (SGLT1の場合)又は 400 μ L (SGLT2の場合)に溶かした。液体シ ンチレーター(パーキンエルマ一社)を加えよく混和した後、 microBETA (SGLTlの 場合)又は液体シンチレーシヨンカウンター(SGLT2の場合)(ベックマンコールター 社)で放射活性を測定した。対照群として試験化合物を含まな!/ヽ取り込み用緩衝液 を調製した。また基礎取り込み用として NaClに代えて塩ィ匕コリンを含む取り込み用緩 衝液を調製した。 ) Wash twice with 200 (for 301/11) or 2111 (for 301 ^ 2) and add to 0.2 M NaO H solution 75 μL (for SGLT1) or 400 μL (for SGLT2) Melted. After adding a liquid scintillator (Perkin Elma) and mixing well, the radioactivity was measured with a microBETA (for SGLTl) or a liquid scintillation counter (for SGLT2) (Beckman Coulter). As a control group, a test compound-free buffer was prepared. In addition, a buffer solution for uptake containing salty choline instead of NaCl was prepared for basic uptake.
[0150] IC 値を求めるにあたり、適当な 6濃度の試験化合物を用い、対照群の糖取り込み [0150] In determining the IC value, the appropriate 6 concentrations of test compound were used, and the glucose uptake of the control group
50 50
量(100%)に対し、糖取り込み量が 50%阻害される試験化合物濃度 (IC 値)を算 Calculate the concentration (IC value) of the test compound that inhibits the sugar uptake by 50% against the amount (100%).
50 出した。試験結果を表 4に示す。 50 out. Table 4 shows the test results.
[0151] [表 4]
IC50 for に 50 for [0151] [Table 4] IC50 for to 50 for
化合物番号 Compound number
hSGLTI (nM) hSGLT2 (nM) hSGLTI (nM) hSGLT2 (nM)
1 205 ― 1 205 ―
2 79 > 100,000 2 79> 100,000
3 57 > 100,000 3 57> 100,000
4 96 > 10,000 4 96> 10,000
5 15 >100,000 5 15> 100,000
6 66 > 100,000 6 66> 100,000
7 21 > 100,000 7 21> 100,000
8 382 ― 8 382 ―
9 42 > 100,000 9 42> 100,000
10 332 ― 10 332 ―
1 1 164 一 1 1 164 1
12 159 一 12 159
13 14 1991 13 14 1991
14 22 761 3 14 22 761 3
15 15 597 15 15 597
16 28 2314 16 28 2314
17 34 4393 17 34 4393
18 18 567 試験例 2 18 18 567 Test Example 2
ストレブトゾトシン糖尿病モデルラットにおける血糖値上昇抑制作用確認試験 (1)糖尿病モデルラットの作製 Test for confirming inhibitory effect on blood glucose level in streptozotocin diabetic model rats (1) Preparation of diabetic model rats
7週齢の SDZIGSラット(日本チヤ一ルスリバ一株式会社,雄性)について約 16時 間の絶食後、エーテル麻酔下でストレブトゾトシン (STZ) 50mgZkgを尾静脈内投
与し、糖尿病モデルラットを作製した.同様にエーテル麻酔下, 1. 25mmolZLタエ ン酸生理食塩液 lmLZkgを尾静脈内投与し、正常対照ラットを作製した。 STZまた は 1. 25mmolZLクェン酸生理食塩液投与 1週後(8週齢)、経口グルコース負荷試 験に供した。 Seven-week-old SDZIGS rats (Nippon Chilli Sliver Co., Ltd., male) were fasted for about 16 hours and then injected with streptozotocin (STZ) 50 mgZkg into the tail vein under ether anesthesia. A diabetic model rat was prepared in the same manner, and under ether anesthesia, 1.25 mmol ZL taenoic acid physiological saline lmLZkg was administered into the tail vein to prepare a normal control rat. One week after administration of STZ or 1.25 mmol ZL citrate physiological saline (8 weeks old), it was subjected to an oral glucose tolerance test.
(2)経口グルコース負荷試験 (2) Oral glucose tolerance test
ラットを約 16時間の絶食後、薬物投与群には、 0. 5%カルボキシメチルセルロース (CMC)水溶液に懸濁した薬物(lmgZkg)を,対照群には 0. 5%CMC水溶液の み経口投与した。薬物投与 5分後に、グルコース溶液 (2gZkg)を経口投与し、薬物 投与前 (Otime)、及び、経口投与 0. 25、 0. 5、 1, 2時間後の計 5点で採血した。 After fasting the rats for about 16 hours, the drug group was orally administered with a drug (lmgZkg) suspended in a 0.5% carboxymethylcellulose (CMC) aqueous solution and only a 0.5% CMC aqueous solution in the control group. . Five minutes after drug administration, glucose solution (2 gZkg) was orally administered, and blood was collected at a total of 5 points before drug administration (Otime) and 0.25, 0.5, 1, 2 hours after oral administration.
[0153] 採血は、エーテル麻酔下でラット眼窩静脈洞よりへノ^ンコート採血管を用いて行 い、遠心分離後、血漿を分取した。血漿中グルコース濃度の定量は、グルコース CII テストヮコー (和光純薬株式会社)を用いて測定した。血糖値上昇抑制作用強度は、 各薬物投与群の 0から 1時間までの血糖値より台形法を用いて血糖値-時間曲線下 面積 (AUC)を算出し、 basalを差し引いた血糖増加面積 ( AAUC)として表記し、対照 群のそれに対する降下の割合で表記した。結果を表 5に示す。 [0153] Blood was collected from the orbital sinus of the rat under ether anesthesia using a non-coated blood collection tube, centrifuged, and plasma was collected. The plasma glucose concentration was measured using Glucose CII Test Sakai (Wako Pure Chemical Industries, Ltd.). The increase in blood glucose level is calculated by calculating the area under the blood glucose level-time curve (AUC) using the trapezoidal method from the blood glucose level from 0 to 1 hour in each drug administration group, and subtracting basal (AAUC) ) And expressed as the rate of descent relative to that of the control group. The results are shown in Table 5.
[0154] [表 5] [0154] [Table 5]

本発明により、小腸上皮に発現する SGLT1 (ナトリウム依存性グルコース共輸送体
1)を選択的に阻害し、小腸力もの糖吸収を阻害することによって、 IGT (耐糖能異常 )を制御するピラゾリル 5—チォダルコシドィヒ合物を有効成分として含有する糖尿病 の予防又は治療剤を提供することが期待される。
According to the present invention, SGLT1 (sodium-dependent glucose cotransporter expressed in the intestinal epithelium) Diabetes prevention or treatment containing pyrazolyl 5-thiodarcosidhi compound as an active ingredient by selectively inhibiting 1) and inhibiting sugar absorption of small intestine by controlling IGT (abnormal glucose tolerance) It is expected to provide an agent.
Claims
[化 1] [Chemical 1]

式中、 Zは、 C アルキル基であり、 Where Z is a C alkyl group,
1-6 1-6
アルコキシ基
 Alkoxy group
Alkoxy group
または C アルキル基であり、 Or a C alkyl group,
1-6 1-6
Yは単結合、 C アルキレン基、 C ァルケ-レン基または— O— (CH ) n— (nは 1 Y is a single bond, a C alkylene group, a C alkylene group or —O— (CH) n— (n is 1
1-6 2-6 2 1-6 2-6 2
から 4の整数を示す)であり、 Represents an integer from 4 to 4)
Qは水酸基、 - OC アルキルフエ-ル、—N(RA)RBまたは CONHRCである、 Q is a hydroxyl group, -OC alkyl file, -N (R A ) R B or CONHRC,
1-4 1-4
但し、 Qが水酸基、 - OC アルキルフエ-ルまたは— NHのとき、 Yは単結合で However, when Q is a hydroxyl group, -OC alkylphenol or -NH, Y is a single bond.
1-4 2 1-4 2
はない。 There is no.
RA及び RBは同一または異なって、水素原子、—CONHRD (RDは水素原子ま たは 、水酸基及びピリジル基からなる群から選択される置換基で置換されてもよ!ヽ C ァ R A and R B may be the same or different and may be substituted with a hydrogen atom, —CONHR D (R D is a hydrogen atom or a substituent selected from the group consisting of a hydroxyl group and a pyridyl group!
1 - 6 ルキル基を示す)、 C (二 NH) NHRE (REは水素原子、または— CO C アルキル 1-6 alkyl), C (2 NH) NHR E (R E is a hydrogen atom, or —CO C alkyl
2 1-4 フエニルである)、あるいは、 C アルキル基 (該 C アルキル基は 、水酸基、 CO 2 1-4 phenyl) or a C alkyl group (the C alkyl group is a hydroxyl group, CO
1-6 1-6 1-6 1-6
NH及び OC アルキルフエニルからなる群から選択される置 換基で置換されて Substituted with a substituent selected from the group consisting of NH and OC alkylphenyl.
2 1-4 2 1-4
もよい)であり、 Is good)
は水酸基及び— CON (RF) 力もなる群より選択される置換基で置換された C Is C substituted with a substituent selected from the group consisting of a hydroxyl group and a —CON (R F ) force
1- アルキル基であり、 1-alkyl group,
6 6
RF及び Rは、同一または異なって、水素原子または C アルキル基である力 ある
いは RF及び Rが結合している窒素原子と一緒になつて、さらに環構成原子として、 酸素原子、硫黄原子及び窒素原子から選択されるへテロ原子を含んでも良い 5〜6 員のへテロシクロアルキル基 (該ヘテロシクロアルキル基は水酸基で置換されても良R F and R are the same or different and may be a hydrogen atom or a C alkyl group There are R F and R a together with the nitrogen atom to which they are attached connexion, a further ring-constituting atom, an oxygen atom, a sulfur atom and to the to be good 5-6 membered comprise hetero atoms selected from nitrogen atom Telocycloalkyl group (the heterocycloalkyl group may be substituted with a hydroxyl group)
V、C アルキル基で置換されてもょ 、)を形成してもよ!/、。 V, C may be substituted with alkyl groups, may form)! / ,.
1-6 1-6
[2] Yが C アルキレン基、 C アルケニレン基または一 O— (CH ) n— (nは 2 [2] Y is a C alkylene group, C alkenylene group or one O— (CH) n— (n is 2
1-6 2-6 2 1-6 2-6 2
力も 4の整数である)である、請求項 1記載のピラゾリル 5 チォダルコシド化合物若 しくはその製薬学的に許容される塩又はそれらの水和物。 The pyrazolyl 5 thiodarcoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof according to claim 1, wherein the force is an integer of 4.
[3] Qが— N(RA)RB (RA及び RBは請求項 1で定義したとおりである)である、請求項 1ま たは 2に記載のピラゾリル 5 チォダルコシド化合物若しくはその製薬学的に許容さ れる塩又はそれらの水和物。 [3] The pyrazolyl 5 thiodarcoside compound or the pharmaceutical thereof according to claim 1 or 2, wherein Q is —N (R A ) R B (R A and R B are as defined in claim 1). Pharmaceutically acceptable salt or hydrate thereof.
[4] RAが― CONHRD (RDは水酸基及びピリジル基からなる群から選択される置換基で 置換されてもょ 、C アルキル基である)であり、 RBが水素原子である、請求項 3に記 [4] R A is —CONHR D (R D is a C alkyl group which may be substituted with a substituent selected from the group consisting of a hydroxyl group and a pyridyl group), and R B is a hydrogen atom. Claim 3
1-6 1-6
載のピラゾリル 5—チォダルコシドィ匕合物若しくはその製薬学的に許容される塩又 はそれらの水和物。 Pyrazolyl 5-thiodarcoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof.
[5] RAが— C ( = NH) NHRE(REは水素原子、または— CO C アルキルフエ-ルであ [5] R A is — C (= NH) NHR E (R E is a hydrogen atom or — CO C alkylphenol)
2 1-4 2 1-4
る)であり、 RBが水素原子である、請求項 3に記載のピラゾリル 5—チォダルコシドィ匕 合物若しくはその製薬学的に許容される塩又はそれらの水和物。 4. The pyrazolyl 5-thiodarcoside compound or the pharmaceutically acceptable salt thereof or the hydrate thereof according to claim 3, wherein R B is a hydrogen atom.
[6] RA及び RBが同一または異なって、水素原子、または C アルキル基 (該 C アルキ [6] R A and R B are the same or different and each represents a hydrogen atom or a C alkyl group (the C alkyl group).
1-6 1-6 ル基は、水酸基、 CONH及び OC アルキルフヱニルからなる群から選択され 1-6 1-6 group is selected from the group consisting of hydroxyl, CONH and OC alkylphenyl.
2 1-4 2 1-4
る置換基で置換されてもょ 、)である請求項 3記載のピラゾリル 5—チォダルコシド 化合物若しくはその製薬学的に許容される塩又はそれらの水和物。 The pyrazolyl 5-thiodarcoside compound according to claim 3, or a pharmaceutically acceptable salt thereof, or a hydrate thereof.
[7] Qが— CONHRe ( は請求項 1で定義したとおりである)である、請求項 1または 2 に記載のピラゾリル 5—チォダルコシドィ匕合物若しくはその製薬学的に許容される 塩又はそれらの水和物。 [7] The pyrazolyl 5-thiodalcoside compound or the pharmaceutically acceptable salt thereof or the pharmaceutically acceptable salt thereof according to claim 1 or 2, wherein Q is —CONHR e (is as defined in claim 1). Hydrate.
[8] が水酸基及び— CONH力もなる群より選択される 1〜4個の置換基で置換され [8] is substituted with 1 to 4 substituents selected from the group consisting of a hydroxyl group and —CONH force.
2 2
たじ アルキル基である力 または、 CO ピペラジノ (該ピペラジノは、水酸基で置 The force of an alkyl group or CO piperazino (the piperazino is a hydroxyl group
1-6 1-6
換されても良い C アルキル基で置換されてもよい)で置換された C アルキル基で A C alkyl group substituted with a C alkyl group which may be substituted)
1-6 1-6 1-6 1-6
ある(ここで、 RGがー NH—に結合する各 C アルキル基の炭素原子は 3級である)、
請求項 7記載のピラゾリル 5—チォダルコシド化合物若しくはその製薬学的に許容 される塩又はそれらの水和物。 (Wherein the carbon atom of each C alkyl group where R G is bonded to —NH— is tertiary), The pyrazolyl 5-thiodarcoside compound according to claim 7, or a pharmaceutically acceptable salt thereof, or a hydrate thereof.
[9] Yは C アルキレン基又は C ァルケ-レン基である、請求項 7または 8記載のビラ [9] The blade according to claim 7 or 8, wherein Y is a C alkylene group or a C alkylene group.
1-6 2-6 1-6 2-6
ゾリル 5—チォダルコシド化合物若しくはその製薬学的に許容される塩又はそれら の水和物。 Zolyl 5-thiodarcoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof.
[10] アルキル基であり、
 [10] an alkyl group,
[10] an alkyl group,
Yは、— O— (CH ) n- (nは 1から 4の整数である)であり、 Y is —O— (CH 2) n- (n is an integer from 1 to 4);
2 2
Qは、水酸基、— OC アルキルフエニル、又は— N(RA)RBであり、 Q is a hydroxyl group, —OC alkylphenyl, or —N (R A ) R B ;
1-4 1-4
RA及び RBは、同一又は異なって、水素原子、 -CONHで置換された C アルキ R A and R B are the same or different and are each a hydrogen atom, C alkyl substituted with -CONH.
2 1-6 ル基、— CONHRD (RDは、水酸基で置換された C アルキル基である)、あるいは— 2 1-6 group, — CONHR D (R D is a C alkyl group substituted with a hydroxyl group), or —
1-6 1-6
C (二 NH) NHRE (REは、水素原子または— CO C アルキルフエニルである)である C (2 NH) NHR E (R E is a hydrogen atom or —CO C alkylphenyl)
2 1-4 2 1-4
、請求項 1記載のピラゾリル 5—チォダルコシド化合物若しくはその製薬学的に許容 される塩又はそれらの水和物。 The pyrazolyl 5-thiodarcoside compound according to claim 1, or a pharmaceutically acceptable salt thereof, or a hydrate thereof.
[11] 請求項 1〜: L0のいずれか 1項に記載のピラゾリル 5—チォダルコシドィ匕合物若しく はその製薬学的に許容される塩又はそれらの水和物を有効成分として含む、 SGLT 1活性阻害剤。 [11] Claims 1 to: SGLT 1 comprising as an active ingredient the pyrazolyl 5-thiodarcosidyl compound according to any one of L0 or a pharmaceutically acceptable salt thereof or a hydrate thereof. Activity inhibitor.
[12] 請求項 1〜: L0のいずれか 1項に記載のピラゾリル 5—チォダルコシドィ匕合物若しく はその製薬学的に許容される塩又はそれらの水和物を有効成分として含む、糖尿病 の予防又は治療剤。
[12] Claims 1 to: The pyrazolyl 5-thiodarcosidyl compound according to any one of L0 or a pharmaceutically acceptable salt thereof or a hydrate thereof as an active ingredient. Prophylactic or therapeutic agent.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006-128679 | 2006-05-02 | ||
| JP2006128679A JP2009167103A (en) | 2006-05-02 | 2006-05-02 | Pyrazolyl 5-thioglycoside compound |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007129668A1 true WO2007129668A1 (en) | 2007-11-15 |
Family
ID=38667776
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/059386 WO2007129668A1 (en) | 2006-05-02 | 2007-05-02 | Pyrazolyl 5-thioglucoside compound |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2009167103A (en) |
| WO (1) | WO2007129668A1 (en) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| US8080580B2 (en) | 2008-08-28 | 2011-12-20 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8669380B2 (en) | 2009-11-02 | 2014-03-11 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| CN110054657A (en) * | 2018-01-18 | 2019-07-26 | 亚宝药业集团股份有限公司 | Sugar-substituted pyrazole compound of glucopyra and preparation method thereof |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004014932A1 (en) * | 2002-08-08 | 2004-02-19 | Kissei Pharmaceutical Co., Ltd. | Pyrazole derivative, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004018491A1 (en) * | 2002-08-23 | 2004-03-04 | Kissei Pharmaceutical Co., Ltd. | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004019958A1 (en) * | 2002-08-27 | 2004-03-11 | Kissei Pharmaceutical Co., Ltd. | Pyrazole derivatives, medicinal composition containing the same, and medicinal use thereof |
| WO2004031203A1 (en) * | 2002-10-04 | 2004-04-15 | Kissei Pharmaceutical Co., Ltd. | Pyrazole derivative, medicinal composition containing the same, medicinal use thereof and intermediate in producing the same |
| EP1609799A1 (en) * | 2003-04-01 | 2005-12-28 | Taisho Pharmaceutical Co., Ltd | Heteroaryl 5-thio-beta-d-glucopyranoside derivatives and remedies for diabetes containing the same |
| JP2006117651A (en) * | 2004-09-27 | 2006-05-11 | Taisho Pharmaceut Co Ltd | Sglt2 activity inhibitor |
-
2006
- 2006-05-02 JP JP2006128679A patent/JP2009167103A/en active Pending
-
2007
- 2007-05-02 WO PCT/JP2007/059386 patent/WO2007129668A1/en active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004014932A1 (en) * | 2002-08-08 | 2004-02-19 | Kissei Pharmaceutical Co., Ltd. | Pyrazole derivative, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004018491A1 (en) * | 2002-08-23 | 2004-03-04 | Kissei Pharmaceutical Co., Ltd. | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004019958A1 (en) * | 2002-08-27 | 2004-03-11 | Kissei Pharmaceutical Co., Ltd. | Pyrazole derivatives, medicinal composition containing the same, and medicinal use thereof |
| WO2004031203A1 (en) * | 2002-10-04 | 2004-04-15 | Kissei Pharmaceutical Co., Ltd. | Pyrazole derivative, medicinal composition containing the same, medicinal use thereof and intermediate in producing the same |
| EP1609799A1 (en) * | 2003-04-01 | 2005-12-28 | Taisho Pharmaceutical Co., Ltd | Heteroaryl 5-thio-beta-d-glucopyranoside derivatives and remedies for diabetes containing the same |
| JP2006117651A (en) * | 2004-09-27 | 2006-05-11 | Taisho Pharmaceut Co Ltd | Sglt2 activity inhibitor |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US8080580B2 (en) | 2008-08-28 | 2011-12-20 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| US8669380B2 (en) | 2009-11-02 | 2014-03-11 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| US9439901B2 (en) | 2009-11-02 | 2016-09-13 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| US9308204B2 (en) | 2009-11-02 | 2016-04-12 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| CN110054657A (en) * | 2018-01-18 | 2019-07-26 | 亚宝药业集团股份有限公司 | Sugar-substituted pyrazole compound of glucopyra and preparation method thereof |
| CN110054657B (en) * | 2018-01-18 | 2021-06-29 | 亚宝药业集团股份有限公司 | Glucopyranosyl substituted pyrazole compound and preparation method thereof |
| US11377465B2 (en) | 2018-01-18 | 2022-07-05 | Yabao Pharmaceutical Group Co., Ltd. | Pyranoglucose-substituted pyrazole compound, preparation method thereof and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009167103A (en) | 2009-07-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007129668A1 (en) | Pyrazolyl 5-thioglucoside compound | |
| JP5067644B2 (en) | C-phenylglycitol compound | |
| CN101657471B (en) | Bicyclic compounds and use as antidiabetics | |
| WO2007126117A1 (en) | Phenyl 5-thioglucoside compound | |
| TWI298071B (en) | ||
| TWI231298B (en) | Glucopyranosyloxypyrazole derivatives and use thereof in medicines | |
| JPWO2008072726A1 (en) | 1-Phenyl 1-thio-D-glucitol derivative | |
| TWI351281B (en) | Nitrogenous fused-ring derivatives,medicinal compo | |
| WO2008001864A1 (en) | C-phenyl 1-thioglucitol compound | |
| US20090318477A1 (en) | Chemical compounds | |
| CN106349201B (en) | The C- glycosides derivatives of optically pure benzyl -4- chlorphenyls | |
| TW200540166A (en) | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof | |
| KR20160029808A (en) | Substituted benzofuranyl and benzoxazolyl compounds and uses thereof | |
| JPWO2004014931A1 (en) | Aryl 5-thio-β-D-glucopyranoside derivative and therapeutic agent for diabetes containing the same | |
| TW200540167A (en) | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof | |
| WO2011094890A1 (en) | Phenylalanine derivatives and their use as non-peptide glp-1 receptor modulators | |
| WO2006087997A1 (en) | 1-SUBSTITUTED-7-(β-D-GLYCOPYRANOSYLOXY)(AZA)INDOLE COMPOUND AND PHARMACEUTICAL CONTAINING THE SAME | |
| CN1980894A (en) | Compounds and compositions as PPAR modulators. | |
| JP2005247834A (en) | Activity inhibitor of sodium-dependent glucose cotransporter 2 | |
| CA3094820A1 (en) | Modulators of g-protein coupled receptors | |
| JP2009120553A (en) | Therapeutic agent for diabetes containing c-phenyl glucitol compound as effective ingredient | |
| CN1980919A (en) | Compounds and compositions as ppar modulators | |
| JP2009107947A (en) | Agent for treating diabetes, containing pyrazolyl 5-thioglucoside compound as active ingredient | |
| JP2009107948A (en) | Agent for treating diabetes, containing phenyl 5-thioglucoside compound as active ingredient | |
| JP2010018611A (en) | Preventive or therapeutic agent for diabetes, diabetes-related diseases or diabetic complications containing 1-phenyl 1-thio-d-glucitol derivative as effective ingredient |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07742821 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07742821 Country of ref document: EP Kind code of ref document: A1 |