WO2006129100A1 - Novel compounds - Google Patents
Novel compounds Download PDFInfo
- Publication number
- WO2006129100A1 WO2006129100A1 PCT/GB2006/002015 GB2006002015W WO2006129100A1 WO 2006129100 A1 WO2006129100 A1 WO 2006129100A1 GB 2006002015 W GB2006002015 W GB 2006002015W WO 2006129100 A1 WO2006129100 A1 WO 2006129100A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- indazol
- ring
- compound
- phenyl
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates to novel bis-anilinopyrimidine compounds which have activity against the spleen tyrosine kinase (Syk kinase), processes for their preparation, pharmaceutically acceptable formulations containing them and their use in therapy.
- Syk kinase spleen tyrosine kinase
- Allergic rhinitis and asthma are diseases associated with hypersensitivity reactions and inflammatory events involving a multitude of cell types including mast cells, eosinophils, T cells and dendritic cells.
- high affinity immunoglobulin receptors for IgE (Fc ⁇ Rl) and IgG (Fc ⁇ RI) become cross-linked and activate downstream processes in mast cells and other cell types leading to the release of pro-inflammatory mediators and airway spasmogens.
- IgE receptor cross-linking by allergen leads to release of mediators including histamine from pre-formed granules, as well as the synthesis and release of newly synthesised lipid mediators including prostaglandins and leukotrienes.
- Syk kinase is a non-receptor linked tyrosine kinase which is important in transducing the downstream cellular signals associated with cross-linking Fc ⁇ Rl and or Fc ⁇ RI receptors, and is positioned early in the signalling cascade.
- the early sequence of Fc ⁇ Rl signalling following allergen cross-linking of receptor-lgE complexes involves first Lyn (a Src family tyrosine kinase) and then Syk kinase.
- Inhibitors of Syk kinase activity would therefore be expected to inhibit all downstream signalling cascades thereby alleviating the immediate allergic response and adverse events initiated by the release of pro-inflammatory mediators and spasmogens (Wong, B., Grossbard, E. B. Payan, D. G & Masuda, E. S. Expert Opin. Investig. Drugs (2004) 13 (7) 743-762).
- Rheumatoid Arthritis is an auto-immune disease affecting approximately 1% of the population. It is characterised by inflammation of articular joints leading to debilitating destruction of bone and cartilage.
- Recent clinical studies with Rituximab, which causes a reversible B cell depletion, (J. CW. Edwards et al 2004, New Eng. J. Med. 350: 2572-2581) have shown that targeting B cell function is an appropriate therapeutic strategy in auto-immune diseases such as RA.
- Clinical benefit correlates with a reduction in auto-reactive antibodies (or Rheumatoid Factor) and these studies suggest that B cell function and indeed auto-antibody production are central to the ongoing pathology in the disease.
- WO 03/057695 (Boehringer lngelheim Pharmaceuticals, Inc.) describes substituted [1 ,6]-naphthyridines that inhibit Syk kinase.
- WO2003/063794, WO2004/014382, WO2005/012294 and WO2005/16893 describes a series of 2,4-pyrimidinediamaine compounds which inhibit Syk kinase, for use in treating autoimmune diseases.
- WO2005/026158 (Novartis AG) describes 2,4-di (hetero)-arylamino-pyrimidine derivatives which have ZAP-70 and/or Syk inhibitory activities.
- WO 04/035604 discloses the structural co-ordinates of the human Syk protein.
- the present invention provides a compound of formula (I):
- R 1 , R 2 and R 3 is each independently selected from hydrogen, halogen, hydroxy, -C 1-6 alkyl, -NR 5 R 6 , -C 1-6 alkylene-NR 5 R 6 , -CN, -C ⁇ alkylene-COaH, C(O)Ci -6 alkoxy, -C 1-2 alkyl substituted by 1 or more fluorine atoms), -C 1-6 alkoxy, -C(O)C 1-6 alkyl, - C(O)NR 5 R 6 , -C(O)NHR 7 , -OCH 2 C(O)NHR 8 , -OCH 2 C(O)OR 5 , -CH 2 C(O)NHR 8 , -NR 5 C(O)R 6 , -SC 1-6 alkyl, -S(O)C 1-6 alkyl, -S(O) 2 C 1-6 alkyl, -CH 2 S(O) 2 C 1-6 alkyl,
- R 1 is hydrogen, halogen or C 1-6 alkyl and R 2 and R 3 are attached to adjacent carbon atoms of the phenyl ring and are joined and together to form, in combination with the carbon atoms on the phenyl group to which they are attached, a 5- or 6-membered heteroaryl or heterocyclic ring ; or
- R 1 is hydrogen, halogen or C 1-6 alkyl and R 2 and R 3 are attached to adjacent carbon atoms of the phenyl ring and are joined together to form, in combination with the carbon atoms on the phenyl group to which they are attached, a 5- or 6-membered carbocyclic ring, and
- R 7 is phenyl optionally substituted by halogen, C 1-6 alkyl
- R 8 is hydrogen, C 1-6 alkyl optionally terminally substituted with hydroxy, 2-oxo-1- pyrrolidinyl or 2,5-dioxo-1-pyrrolidinyl, a 5- or 6-membered heteroaryl ring comprising 1 or 2 heteroatoms independently selected from O, N and S and optionally substituted on a ring carbon by C 1-6 alkyl, phenyl optionally substituted by halo, Crealkyl.
- C 1-6 alkoxy or trifluoromethyl, or, together with the nitrogen to which it is attached is a C ⁇ alkyl ester of serine;
- R 9 is Ci -6 alkyl or phenyl optionally substituted by C-,. 6 alkyl;
- R 10 is Ci -6 alkoxy, Ci -6 alkyl optionally terminally substituted with hydroxyl, 2-oxo-1- pyrrolidinyl or 2,5-dioxo-1-pyrrolidinyl, or R 10 is a 5 or 6-membered heteroaryl ring comprising 1 heteroatom selected from O, N and S or 2 different heteroatoms independently selected from O, N and S and optionally substituted on one or more ring carbons by C 1-6 alkyl;
- a salt or solvate preferably a pharmaceutically acceptable salt or solvate, thereof.
- the present invention provides for a compound of formula (I) which is of the formula (Ia):
- R 1 , R 2a and R 3a is each independently selected from the group of substituents Group A which comprises hydrogen, halogen, hydroxy, -C 1-6 alkyl, -NR 5 R 6 , -Ci. 6 alkylene-NR 5 R 6 , -CN, -Co -3 alkylene-C0 2 H, C(O)C 1-6 alkoxy, -Ci -2 alkyl substituted by 1 or more fluorine atoms, -C 1-6 alkoxy, -CCOJC L ⁇ alkyl, -C(O)NR 5 R 6 , -C(O)NHR 7 , -OCH 2 C(O)NHR 8 , -OCH 2 C(O)OR 5 , -CH 2 C(O)NHR 8 , -NR 5 C(O)R 6 , -SC 1- ⁇ alkyl, -S(O)C 1-6 alkyl, -S(O) 2 C 1-6 alkyl, -
- R 5 and R 6 are is each independently hydrogen or C 1-6 alkyl
- R 7 , R 8 , R 9 and R 10 are as hereinbefore defined for formula (I); or
- a salt or solvate preferably a pharmaceutically acceptable salt or solvate, thereof.
- Compounds of the present invention are useful as inhibitors of Syk and thus useful in treating diseases resulting from in appropriate mast cell activation, for instance allergic and inflammatory diseases.
- two of R 1 , R 2 and R 3 are hydrogen.
- representative values for substituents in Group A include: hydroxy, chloro, methyl, (1-methylethyl), -CONH 2 , -CONHCH 3 , -CONH(C 2 H 5 ), -CON(C 2 Hs) 2 , -CN, -CF 3 , methyloxy, (i-methylethyl)oxy, -CO 2 H, -CH 2 CO 2 H, - C(O)OCH3,-SO 2 NH 2 , and -SO 2 CH 3 .
- R 1 and R 3 is each hydrogen and R 2 is R 4 .
- R 1 and R 2 is each hydrogen and R 3 is R 4 .
- R 4 include: benzimidazol-2-yl, tetrazol-5-yl ;
- representative examples of the 5- or 6- membered saturated or unsaturated ring formed by R 2a and R 3a , fused with the phenyl ring include:
- representative examples of the 5- or 6- membered saturated or unsaturated ring formed by R 2a and R 3a , fused with the phenyl ring, and including any substituents which may be present, include:
- representative examples of the 5- or 6- membered saturated carbocyclic ring formed by R 2a and R 3a include:
- R 5 and R 6 include: H, methyl, and ethyl.
- R 7 include phenyl and 4- methyl phenyl.
- R 8 include: hydrogen, 3- hydroxypropyl, 2,5-dioxo-1-pyrrolidinyl, 2-oxo-1-pyrrolidinyl, difluoromethyloxy, methyl serinate (including N);
- Representative examples of compounds of formula (I) include:
- heteroaryl includes single or fused rings comprising up to four hetero-atoms in the ring selected from oxygen, nitrogen and sulphur and optionally substituted with up to three substituents.
- Representative heteroaryl rings comprise from 4 to 7, preferably 5 or 6, ring atoms.
- a fused heteroaryl ring system may include carbocyclic rings and need only include one heterocyclic ring.
- heterocyclic includes non-aromatic single or fused rings comprising up to four hetero-atoms in the ring selected from oxygen, nitrogen and sulphur and optionally substituted with up to three substituents.
- Representative heterocyclic rings comprise from 4 to 7, preferably 5 to 6, ring atoms.
- a fused heterocyclic ring system may include carbocyclic rings and need only include one heterocyclic ring.
- Examples of "-C 1-2 alkyl substituted by 1 or more fluorine atoms” include, but is not restricted to, -CF 3 .
- references to alkyl include references to both straight chain and branched chain aliphatic isomers of the corresponding alkyl. It will be appreciated that references to alkylene and alkoxy shall be interpreted similarly.
- references to C 3-7 cycloalkyl include references to all alicyclic (including branched) isomers of the corresponding alkyl.
- carbocyclic refers to rings which may be saturated or unsaturated but which are not aromatic, may be branched, which contain 5-7 carbon atoms and which may be optionally substituted with up to three substituents.
- substituents for such carbocyclic rings include those previously mentioned for heteroaryl and heterocyclic.
- the term “pharmaceutically acceptable” refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salts of the compound of the present invention may be prepared.
- pharmaceutically acceptable salts refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects.
- compositions may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively. Indeed, in certain embodiments of the invention, pharmaceutically acceptable salts may be preferred over the respective free base or free acid because such salts impart greater stability or solubility to the molecule thereby facilitating formulation into a dosage form.
- the compound of the present invention may contain one or more acidic functional groups.
- suitable pharmaceutically acceptable salts include salts of such acidic functional groups.
- Representative salts include pharmaceutically acceptable metal salts such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc salts; carbonates and bicarbonates of a pharmaceutically- acceptable metal cation such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc; pharmaceutically acceptable organic primary, secondary, and tertiary amines including aliphatic amines, aromatic amines, aliphatic diamines, and hydroxy alkylamines such as methylamine, ethylamine, 2-hydroxyethylamine, diethylamine, triethylamine, ethylenediamine, ethanolamine, diethanolamine, and cyclohexylamine.
- Compounds of the present invention are basic and accordingly generally capable of forming pharmaceutically acceptable acid addition salts by treatment with a suitable acid.
- Suitable acids include pharmaceutically acceptable inorganic acids and pharmaceutically acceptable organic acids.
- Representative pharmaceutically acceptable acid addition salts include hydrochloride, hydrobromide, nitrate, methylnitrate, sulfate, bisulfate, sulfamate, phosphate ⁇ , acetate, hydroxyacetate, phenylacetate, propionate, butyrate, isobutyrate, valerate, maleate, hydroxymaleate, acrylate, fumarate, malate, tartrate, citrate, salicylate, p-aminosalicyclate, glycollate, lactate, heptanoate, phthalate, oxalate, succinate, benzoate, o-acetoxybenzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, mandelate,
- the term "compound” refers to one or more compounds.
- a compound of the present invention refers to one or more compounds of the present invention.
- the compound of the present invention may exist in solid or liquid form. In the solid state, the compound of the present invention may exist in crystalline or noncrystalline form, or as a mixture thereof.
- pharmaceutically acceptable solvates may be formed wherein solvent molecules are incorporated into the crystalline lattice during crystallization.
- Solvates may involve non-aqueous solvents such as, but not limited to, ethanol, isopropanol, DMSO, acetic acid, ethanolamine, and ethyl acetate, or they may involve water as the solvent that is incorporated into the crystalline lattice. Solvates wherein water is the solvent incorporated into the crystalline lattice are typically referred to as "hydrates.” Hydrates include stoichiometric hydrates as well as compositions containing variable amounts of water. The invention includes all such solvates.
- polymorphs may exhibit polymorphism (i.e. the capacity to occur in different crystalline structures). These different crystalline forms are typically known as "polymorphs.”
- the invention includes all such polymorphs. Polymorphs have the same chemical composition but differ in packing, geometrical arangement, and other descriptive properties of the crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deformability, stability, and dissolution properties. Polymorphs typically exhibit different melting points, IR spectra, and X-ray powder diffraction patterns, which may be used for identification.
- polymorphs may be produced, for example, by changing or adjusting the reaction conditions or reagents, used in making the compound. For example, changes in temperature, pressure, or solvent may result in polymophs. In addition, one polymorph may spontaneously convert to another polymorph under certain conditions.
- the compound of the present invention may contain one or more asymmetric centers (also referred to as a chiral center) and may, therefore, exist as individual enantiomers, diastereoisomers, or other stereoisomeric forms, or as mixtures thereof.
- Chiral centers such as chiral carbon atoms, may also be present in a substituent such as an alkyl group. Where the stereochemistry of a chiral center is not specified the structure is intended to encompass any stereoisomer and all mixtures thereof.
- the compound of the present invention containing one or more chiral centers may be used as racemic mixtures, enantiomerically enriched mixtures, or as enantiomerically pure individual stereoisomers.
- Individual stereoisomers of a compound according to Formula (I) which contain one or more asymmetric center may be resolved by methods known to those skilled in the art. For example, such resolution may be carried out (1) by formation of diastereoisomeric salts, complexes or other derivatives; (2) by selective reaction with a stereoisomer-specific reagent, for example by enzymatic oxidation or reduction; or (3) by gas-liquid or liquid chromatography in a chiral enviornment, for example, on a chiral support such as silica with a bound chiral ligand or in the presence of a chiral solvent.
- stereoisomers may be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
- the compound of the present invention may also contain double bonds or other centers of geometric asymmetry. Where the stereochemistry of a centre of geometric asymmetry is not specified, the structure is intended to encompass the trans (E) geometric isomer, the cis (Z) geometric isomer, and all mixtures thereof. Likewise, all tautomeric forms are also included the compound of the present invention whether such tautomers exist in equilibrium or predominately in one form.
- the present invention provides a process for preparing a compound of formula (I), or a salt or solvate thereof, which process comprises:
- Process (a) may be performed in the presence of a solvent (for example, aqueous acetone) and optionally in the presence of dilute acid such as hydrochloric acid at a suitable temperature, preferably in the range of 0-100° C.
- a solvent for example, aqueous acetone
- dilute acid such as hydrochloric acid
- suitable temperature preferably in the range of 0-100° C.
- suitable leaving groups (L 1 ) include, but are not restricted to, an alky!
- alkyl and aryl sulfide an alkyl and aryl sulfinyl, an alkyl and aryl sulfonyl, a halide (such as chloride, bromide or iodide), and an alcohol derived leaving group (such as triflate, mesylate, methylsulfonate or tosylate).
- a halide such as chloride, bromide or iodide
- an alcohol derived leaving group such as triflate, mesylate, methylsulfonate or tosylate.
- process (a) may be performed in the presence of a non-nucleophilic base such as triethylamine or diisopropylethylamine, as a melt, at a suitable temperature, preferably in the range of 0-180° C.
- a non-nucleophilic base such as triethylamine or diisopropylethylamine
- Process (b) may be performed under conditions analogous to those described for process (a) above. Suitable leaving groups for L 2 are also described above for L 1 .
- Suitable amine protecting groups include, but are not restricted to, sulphonyl (such as tosyl), acyl (such as benzyloxycarbonyl or t-butoxycarbonyl) and arylalkyl (such as benzyl), which may be removed by hydrolysis or hydrogenolysis as appropriate.
- Suitable amine protecting groups include trifluoroacetyl (-C(O)CF 3 ), which may be removed by base catalysed hydrolysis, or a solid phase resin bound benzyl group, such as a Merrifield resin bound 2,6-dimethoxybenzyl group (Ellman linker) which may be removed by acid catalysed hydrolysis (using, for example, trifluoroacetic acid).
- a solid phase resin bound benzyl group such as a Merrifield resin bound 2,6-dimethoxybenzyl group (Ellman linker) which may be removed by acid catalysed hydrolysis (using, for example, trifluoroacetic acid).
- Compounds of formula (II) may be prepared according to a process comprising: (a) reacting a compound of formula (Vl):
- L 1 and L 3 represent leaving groups, with a compound of formula (V) as defined above.
- Process (b) may be performed under analogous conditions as those described above for part (a).
- the product of this particular process is advantageously purified by methods well known to persons skilled in the art, such as flash chromatography or high performance liquid chromatography.
- Suitable leaving groups L 3 include those mentioned above for L 1 , especially halide.
- Reduction may be performed using reagents such as, but not limited to, hydrogen gas over palladium, iron metal with hydrochloric acid, stannous chloride and hydrochloric acid, or sodium dithionite, as appropriate under conventional conditions.
- reagents such as, but not limited to, hydrogen gas over palladium, iron metal with hydrochloric acid, stannous chloride and hydrochloric acid, or sodium dithionite, as appropriate under conventional conditions.
- Compounds of formula (VIII) may be prepared by reacting an organic nitrite derivative, such as tert-butyl nitrite, with a compound of formula (IX):
- This reaction may be performed in the presence of an acid, such as acetic acid or hydrochloric acid.
- Certain compounds of formula (IV) may be prepared by a process comprising reacting a compound of formula (VII) or a protected derivative thereof, wherein L 1 and L 3 represent suitable leaving groups, with a compound of formula (111) or a protected derivative thereof.
- This process may be performed under basic conditions, such as tert-butanol and potassium carbonate, and advantageously at a temperature in the range of 50 to 150 0 C.
- Compounds of formula (IV) may also be prepared from an analogous compound where the group L 2 is a hydroxyl group (which can be converted to a halogen by reaction with POCI 3 or PCI 5 ) or a methylthio group (which can be converted to a methylsulfinyl or methylsulfonyl group by oxidation with metachloroperoxybenzoic acid or ozone, for example).
- Compounds in which L 2 is a hydroxyl group may be prepared from corresponding compounds in which L is a methylthio group by oxidation with hydrogen peroxide in acetic acid.
- reaction is preferably performed in the presence of an acid, such as acetic acid or hydrochloric acid.
- the aforementioned reaction may be performed under nitrogen atom protecting conditions (for example, in the presence of acetate) to yield a product in which an indazolyl nitrogen atom is protected.
- Compounds of formula (X) may be prepared by reducing the corresponding nitro compound using conventional conditions, such as sodium dithionite, hydrogen gas in the presence of palladium, or iron metal with hydrochloric acid. Such corresponding nitro compounds may be prepared by reacting a compound of formula (VII) with a compound of formula (Xl):
- Compounds of the present invention are useful as inhibitors of Syk and thus useful in treating diseases resulting from inappropriate mast cell activation, for instance allergic and inflammatory diseases.
- the present invention provides for a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, for use in therapy.
- the present invention provides for a method of treating inappropriate mast cell activation which method comprises administering to a patient in need thereof an effective compound of formula I, or a or a pharmaceutically acceptable salt or solvate thereof.
- the present invention provides a method comprising administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, to inhibit a Syk kinase.
- the present invention provides a method of treating an inflammatory disease which comprises administering to a patient in need thereof an effective compound of formula I, or a pharmaceutically acceptable salt or solvate thereof.
- the present invention provides a method of treating an allergic disorder which comprises administering to a patient in need thereof an effective compound of formula I, or a pharmaceutically acceptable salt or solvate thereof.
- Syk kinase diseases and pathological conditions thought to be mediated by Syk kinase include inflammatory and allergic disorders involving mast cell activation, such as chronic obstructive pulmonary disease (COPD), adult respiratory distress syndrome (ARDS), asthma, ulcerative colitis, Crohn's Disease, bronchitis, conjunctivitis, psoriasis, sclerodoma, urticaria, dermatitis, and allergic rhinitis. They also include inflammatory conditions which involve B cells, for instance lupus and rheumatoid arthritis. Likely disease targets for compounds of the present invention include chronic obstructive pulmonary disease (COPD), asthma and allergic rhinitis.
- COPD chronic obstructive pulmonary disease
- ARDS adult respiratory distress syndrome
- ARDS adult respiratory distress syndrome
- ARDS adult respiratory distress syndrome
- ARDS adult respiratory distress syndrome
- ARDS adult respiratory distress syndrome
- ARDS adult respiratory distress syndrome
- ARDS
- the present invention provides a method of treating chronic obstructive pulmonary disease which comprises administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof.
- the present invention provides a method of treating asthma which comprises administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof.
- the present invention provides a method of treating allergic rhinitis which comprises administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof.
- Compounds of the present invention may also be used in combination with other therapeutic agents, for instance an anti-inflammatory steroid, in particular an antiinflammatory corticosteroid.
- Anti-inflammatory corticosteroids are well known in the art. Representative examples include fluticasone propionate (e.g. see US patent 4,335,121), beclomethasone 17- propionate ester, beclomethasone 17,21-dipropionate ester, dexamethasone or an ester thereof, mometasone or an ester thereof (e.g. mometasone furoate), ciclesonide, budesonide, and flunisolide.
- fluticasone propionate e.g. see US patent 4,335,121
- beclomethasone 17- propionate ester beclomethasone 17,21-dipropionate ester
- dexamethasone or an ester thereof dexamethasone or an ester thereof
- mometasone or an ester thereof e.g. mometasone furoate
- ciclesonide e.g. mometasone furoate
- ciclesonide esonide
- anti-inflammatory corticosteroids are described in WO 02/12266 A1 (Glaxo Group Ltd), in particular, the compounds of Example 1 ( 6 ⁇ ,9 ⁇ -difluoro-17 ⁇ -[(2-furanylcarbonyl)oxy]-11 ⁇ -hydroxy- 16 ⁇ -methyl-3-oxo-androsta-1,4-diene-17 ⁇ -carbothioic acid S-fluoromethyl ester) and Example 41 (6 ⁇ ,9 ⁇ -difluoro-11 ⁇ -hydroxy-16 ⁇ -methyl-17 ⁇ -[(4-methyl-1 ,3-thiazole-5- carbonyl)oxy]-3-oxo-androsta-1 ,4-diene-17 ⁇ -carbothioic acid S-fluoromethyl ester), or a pharmaceutically acceptable salt thereof.
- the compound of the present invention will normally, but not necessarily, be formulated into pharmaceutical compositions prior to administration to a patient. Accordingly, in another aspect the invention is directed to pharmaceutical compositions comprising a compound of the invention and one or more pharmaceutically acceptable excipient.
- compositions of the invention may be prepared and packaged in bulk form wherein a safe and effective amount of a compound of the invention can be extracted and then given to the patient, such as with powders or syrups.
- the pharmaceutical compositions of the invention may be prepared and packaged in unit dosage form wherein each physically discrete unit contains a safe and effective amount of a compound of the invention.
- the pharmaceutical compositions of the invention typically contain from about 0.1 to 99.9 wt.%, depending on the nature of the formulation.
- compositions of the invention typically contain one compound of the invention. However, in certain embodiments, the pharmaceutical compositions of the invention contain more than one compound of the invention. For example, in certain embodiments the pharmaceutical compositions of the invention contain two compounds of the invention. In addition, the pharmaceutical compositions of the invention may optionally further comprise one or more additional pharmaceutically active compounds.
- pharmaceutically acceptable excipient means a pharmaceutically acceptable material, composition or vehicle involved in giving form or consistency to the pharmaceutical composition.
- Each excipient must be compatible with the other ingredients of the pharmaceutical composition when commingled, such that interactions which would substantially reduce the efficacy of the compound of the invention when administered to a patient and would result in pharmaceutically unacceptable compositions are avoided.
- each excipient must of course be of sufficiently high purity to render it pharmaceutically acceptable.
- dosage forms include those adapted for (1) inhalation, such as aerosols and solutions and (2) intranasal administration, such as solutions or sprays.
- Suitable pharmaceutically acceptable excipients will vary depending upon the particular dosage form chosen.
- suitable pharmaceutically acceptable excipients may be chosen for a particular function that they may serve in the composition.
- certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage forms.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the carrying or transporting the compound of the present invention once administered to the patient from one organ, or portion of the body, to another organ, or portion of the body.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to enhance patient compliance.
- Suitable pharmaceutically acceptable excipients include the following types of excipients: Diluents, fillers, binders, lubricants, glidants, granulating agents, solvents, co-solvents, suspending agents, emulsifiers, anticaking agents, humectants, chelating agents, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents.
- Diluents Diluents, fillers, binders, lubricants, glidants, granulating agents, solvents, co-solvents, suspending agents, emulsifiers, anticaking agents, humectants, chelating agents, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents.
- certain pharmaceutically acceptable excipients may serve more than one function and may serve alternative functions depending on how much of the excipient is present in the formulation and what other ingredients are
- compositions of the invention are prepared using techniques and methods known to those skilled in the art. Some of the methods commonly used in the art are described in Remington's Pharmaceutical Sciences (Mack Publishing Company).
- Oral solid dosage forms such as tablets will typically comprise one or more pharmaceutically acceptable excipients, which may for example help impart satisfactory processing and compression characteristics, or provide additional desirable physical characteristics to the tablet.
- pharmaceutically acceptable excipients may be selected from diluents, binders, glidants, lubricants, disintegrants, colorants, flavorants, sweetening agents, polymers, waxes or other solubility- modulating materials.
- Dosage forms for parenteral administration will generally comprise fluids, particularly intravenous fluids, i.e., sterile solutions of simple chemicals such as sugars, amino acids or electrolytes, which can be easily carried by the circulatory system and assimilated. Such fluids are typically prepared with water for injection USP. Fluids used commonly for intravenous (IV) use are disclosed in Remington, The Science and Practice of Pharmacy [full citation previously provided]. The pH of such IV fluids may vary, and will typically be from 3.5 to 8 as known in the art. Dosage forms for nasal or inhaled administration may conveniently be formulated as aerosols, solutions, drops, gels or dry powders.
- Dosage forms for nasal administration may be provided in a metered dose device.
- the dosage form may be provided as a fluid formulation for delivery from a fluid dispenser having a dispensing nozzle or dispensing orifice through which a metered dose of the fluid formulation is dispensed upon the application of a user-applied force to a pump mechanism of the fluid dispenser.
- Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid formulation, the doses being dispensable upon sequential pump actuations.
- the dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid formulation into the nasal cavity.
- the fluid dispenser is of the general type described and illustrated in WO-A-2005/044354.
- the dispenser has a housing which houses a fluid discharge device having a compression pump mounted on a container for containing a fluid formulation.
- the housing has at least one finger-operable side lever which is movable inwardly with respect to the housing to cam the container upwardly in the housing to cause the pump to compress and pump a metered dose of the formulation out of a pump stem through a nasal nozzle of the housing.
- a particularly preferred fluid dispenser is of the general type illustrated in Figures 30-40 of WO-A-2005/044354.
- the compound or salt of formula (I) is in a particle-size-reduced form, and more preferably the size-reduced form is obtained or obtainable by micronisation.
- the preferable particle size of the size-reduced (e.g. micronised) compound or salt or solvate is defined by a D50 value of about 0.5 to about 10 microns (for example as measured using laser diffraction).
- Aerosol formulations can comprise a solution or fine suspension of the active substance in a pharmaceutically acceptable aqueous or non-aqueous solvent. Aerosol formulations can be presented in single or multidose quantities in sterile form in a sealed container, which can take the form of a cartridge or refill for use with an atomising device or inhaler. Alternatively the sealed container may be a unitary dispensing device such as a single dose nasal inhaler or an aerosol dispenser fitted with a metering valve (metered dose inhaler) which is intended for disposal once the contents of the container have been exhausted.
- a metering valve metered dose inhaler
- the dosage form comprises an aerosol dispenser
- it preferably contains a suitable propellant under pressure such as compressed air, carbon dioxide or an organic propellant such as a hydrofluorocarbon (HFC).
- suitable HFC propellants include 1 , 1 ,1 , 2,3,3, 3-heptafluoropropane and 1 ,1 ,1 ,2-tetrafluoroethane.
- the aerosol dosage forms can also take the form of a pump-atomiser.
- the pressurised aerosol may contain a solution or a suspension of the active compound. This may require the incorporation of additional excipients e.g. co-solvents and/or surfactants to improve the dispersion characteristics and homogeneity of suspension formulations. Solution formulations may also require the addition of co-solvents such as ethanol.
- Other excipient modifiers may also be incorporated to improve, for example, the stability and/or taste and/or fine particle mass characteristics (amount and/or profile) of the formulation.
- the pharmaceutical composition is a dry powder inhalable composition.
- a dry powder inhalable composition can comprise a powder base such as lactose, glucose, trehalose, mannitol or starch, the compound of formula (I) or salt or solvate thereof (preferably in particle-size-reduced form, e.g. in micronised form), and optionally a performance modifier such as L-leucine or another amino acid, and/or metals salts of stearic acid such as magnesium or calcium stearate.
- the dry powder inhalable composition comprises a dry powder blend of lactose and the compound of formula (I) or salt thereof.
- the lactose is preferably lactose hydrate e.g. lactose monohydrate and/or is preferably inhalation-grade and/or fine-grade lactose.
- the particle size of the lactose is defined by 90% or more (by weight or by volume) of the lactose particles being less than 1000 microns (micrometres) (e.g. 10-1000 microns e.g. 30-1000 microns) in diameter, and/or 50% or more of the lactose particles being less than 500 microns (e.g. 10-500 microns) in diameter. More preferably, the particle size of the lactose is defined by 90% or more of the lactose particles being less than 300 microns (e.g.
- the particle size of the lactose is defined by 90% or more of the lactose particles being less than 100-200 microns in diameter, and/or 50% or more of the lactose particles being less than 40-70 microns in diameter.
- a suitable inhalation-grade lactose is E9334 lactose (10% fines) (Borculo Domo Ingredients, Hanzeplein 25, 8017 JD Zwolle, Netherlands).
- a pharmaceutical composition for inhaled administration can be incorporated into a plurality of sealed dose containers (e.g. containing the dry powder composition) mounted longitudinally in a strip or ribbon inside a suitable inhalation device.
- the container is rupturable or peel-openable on demand and the dose of e.g. the dry powder composition can be administered by inhalation via the device such as the DISKUS TM device, marketed by GlaxoSmithKline.
- the DISKUS TM inhalation device is for example described in GB 2242134 A, and in such a device at least one container for the pharmaceutical composition in powder form (the container or containers preferably being a plurality of sealed dose containers mounted longitudinally in a strip or ribbon) is defined between two members peelably secured to one another; the device comprises: a means of defining an opening station for the said container or containers; a means for peeling the members apart at the opening station to open the container; and an outlet, communicating with the opened container, through which a user can inhale the pharmaceutical composition in powder form from the opened container.
- the compound of the present invention when administered in combination with other therapeutic agents normally administered by the inhaled, intravenous, oral or intranasal route, that the resultant pharmaceutical composition may be administered by the same routes.
- the compound of the present invention may conveniently be administered in amounts of, for example, 1 ⁇ g to 100 mg.
- the precise dose will of course depend on the age and condition of the patient and the particular route of administration chosen.
- Recombinant human Syk was expressed as a His-tagged protein*.
- the activity of Syk was assessed using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay.
- TR-FRET time-resolved fluorescence resonance energy transfer
- the reaction was incubated for 40 minutes at room temperature, then terminated by the addition of 3 ⁇ l of read reagent containing 60 mM EDTA, 15OmM NaCI, 5OnM Streptavidin APC (Prozyme, San Leandro, California, USA), 0.5nM antiphosphotyrosine antibody labelled with W-1024 europium chelate (Wallac OY, Turku, Finland) in 4OmM HEPES pH 7.4. The reaction was further incubated for 60 minutes at room temperature.
- the degree of phosphorylation of Biotin-AAAEEIYGEI was measured using a BMG Rubystar plate reader (BMG LabTechnologies Ltd, Aylesbury, UK) as a ratio of specific 665 nm energy transfer signal to reference europium 620 nm signal.
- the reaction was incubated for 60min at room temperature, then terminated by the addition of 3 ⁇ l of read reagent containing 60 mM EDTA, 15OmM NaCI, 5OnM Streptavidin APC (Prozyme, San Leandro, California, USA), 0.5nM antiphosphotyrosine antibody labelled with W-1024 europium chelate (Wallac OY, Turku, Finland) in 4OmM HEPES pH 7.4. The reaction was further incubated for 45min at room temperature.
- the degree of phosphorylation of Biotin-AAAEEIYGEI was measured using a BMG Rubystar plate reader (BMG LabTechnologies Ltd, Aylesbury, UK) as a ratio of specific 665 nm energy transfer signal to reference europium 620 nm signal.
- Compounds according to the present invention were assayed in this, or a similar Time-resolved fluorescence resonance energy transfer kinase assay, and gave IC 50 values less than 10 ⁇ M.
- the 30OmM Imidazole fractions were pooled buffer exchanged using G25M (Amersham Biosciences, Buckinghamshire, UK) into 2OmM MES pH 6.0, 2OmM NaCI, 1OmM ⁇ McEtOH,10% glycerol.
- the buffer exchanged 6His-Syk was loaded onto a Source15S column (Amersham Biosciences, Buckinghamshire, UK) and the column eluted using a NaCI gradient 0-50OmM over 50 column volumes.
- the 6His-Syk containing fractions were pooled and concentrated by ultra-filtration. The identity of 6His-Syk was confirmed by peptide mass finger printing and intact LC-MS.
- Cells of the mouse fibroblast cell line NIH-3T3 are stably transfected with a cFms- SYK chimera.
- Addition of the ligand (MCSF) produces dimerisation of the chimera resulting in autophosphorylation of the SYK kinase domain.
- MCSF ligand
- Cells are plated at a density of 1x10 5 /well in a volume of 200 ⁇ L growth medium (DMEM containing 10% heat inactivated foetal calf serum, 1% L-glutamine, 400 ⁇ g/mL geneticin and 400 ⁇ g/mL zeocin) in 96 well Collagen 1 coated tissue culture plates. Following incubation at 37 0 C, 10% CO 2 , for 20 hours the cell supernatant is removed and replaced with 200 ⁇ L DMEM containing 1 % penicillin/streptomycin (serum free DMEM). The cells are incubated for one hour under the conditions described above. The medium is removed, 50 ⁇ L appropriately diluted compound solution added and the plate incubated for a further hour.
- DMEM containing 10% heat inactivated foetal calf serum, 1% L-glutamine, 400 ⁇ g/mL geneticin and 400 ⁇ g/mL zeocin
- Cells are stimulated with 25 ⁇ L MCSF (0.66 ⁇ g/mL final) for 20 minutes at 37°C. After removal of the supernatant, the cells are washed with cold PBS and lysed with 100 ⁇ L lysis buffer for 4 hours at 4 0 C.
- cFms ELISA 85 ⁇ L cell lysate is transferred to a 96 well ELISA plate coated with goat anti human M-CSF R capture antibody and incubated for 16 hours at 4°C.
- the plate is washed and a biotinylated anti-phosphotyrosine detection antibody added (100 ⁇ LJwell) for 2 hours at room temperature. This is removed and replaced with 100 ⁇ l_ Streptavidin- HRP for 30minutes.
- Captured phosphorylated SYK is visualised using 100 ⁇ L TMB substrate. The reaction is terminated with 50 ⁇ L 1M sulphuric acid and the absorbance measured at 450nm.
- Compound is prepared as a 1OmM stock in DMSO and a dilution series prepared in DMSO using 9 successive 5-fold dilutions. This dilution series is diluted a further 1 :333 with serum free DMEM to give the concentration range to be tested of 1x10 "5 to 1.54x10 "11 M.
- Compound dilutions are prepared using the Biomek 2000 or Biomek Nx automated robotic pipetting systems.
- the population of B cells observed in this assay are the naive mature IgM/lgD expressing population. These form at least 70% of the purified B cell population (the rest being isotype switched memory B cells) and are the only cells that proliferate as the cells are stimulated with anti-lgM.
- Anti-lgM drives signalling through the B cell receptor which is Syk dependant. Proliferation is a functional measure of B cell signalling that can be measured by observing the incorporation of tritiated methyl thymidine into the cells.
- Purified human tonsillar B cells are resuspended in Buckleys* medium at a concentration of 1.25 x 10 6 ml.
- 160 ⁇ l of cells re-suspended in Buckley's medium is added to the compound and control wells of a 96 well plate.
- the control wells are located on column 11 and 12 of the 96 well plate.
- the background wells are located in column 12 and 20 ⁇ l of 10 ⁇ M control is added to provide an appropriate background control.
- 20 ⁇ l of 1% DMSO is added to the wells in column 11 for the stimulated control.
- the compound titrations are located between columns 1 and 10. Three compounds are run in duplicate on each plate and row A and B are used for the control compound titration.
- the final concentration of DMSO is 0.1% in the assay.
- the cells are left for 45 minutes, after 45 minutes the proliferative stimulus is added to the first 11 wells of the 96 well plate and 20 ⁇ l of medium is added to column 12.
- F(ab')2 fragments of a polyclonal goat anti-sera raised to human IgM is used at a final concentration of 15 ⁇ g/ ml to stimulate the cells. (Biosource. Cat no: AMI 4601 ).
- Tritiated methyl thymidine is added to the cells at a concentration of 1 ⁇ Ci per well. (Amersham, TRK 758). The radioactivity is added 65 hours after the initial stimulus and is left on the cells for 6 to 8 hours. After pulsing with methyl thymidine the cells are harvested on a Skatron 96 well cell harvester onto glass fibre mats. Once these have dried these are counted on a Wallac 1450 Microbeta scintillation counter.
- Data is downloaded as an XL file and IC50's determined using Activity base.
- Buckleys Medium 450 mis Iscoves (Sigma I 3390), 5OmIs FCS, 2.5 g BSA, 5mls Pen/ strep, 5mls Glutamine (20OmM), 500 ⁇ l Apo transferrin (50mg/ml) Sigma (T 1147), 100 ⁇ l Bovine Insulin (10mg/ml) Sigma (1 1882).
- Compound is prepared as a 1OmM stock in DMSO and a dilution series prepared in DMSO using 9 successive 3-fold dilutions.
- This dilution series is diluted a further 1 :100 with Buckleys medium to give the concentration range to be tested of 100 ⁇ M to 5nM.
- This is added as 20 ⁇ l to 96 well plates in duplicate to generate two IC50's for each compound tested. Each plate is run in the presence of a control compound, which acts as an internal standard. .
- LAD2 is a stem cell factor (SCF)-dependent human mast cell line that was established by the NIH from bone marrow aspirates from a patient with mast cell sarcoma/leukaemia.
- SCF stem cell factor
- LAD2 cells resemble CD34+-derived human mast cells and express functional Fc ⁇ Rl.
- the Fc ⁇ RI is up-regulated in the presence of IL-4, SCF and IgE, subsequent cross linking of cell-bound IgE results in degranulation which can be measured as hexosaminidase release.
- LAD2 cells are re-suspended at 1x10 5 /ml in complete stem pro-34SFM (Gibco Cat 10640-019 media containing Stem Pro-34 nutrient supplement (1 :40), glutamine (2mM), penicillin (100u/ml), streptomycin (100 ⁇ g/ml)) with additional supplements of human recombinant SCF (100ng/ml; R&D systems), human recombinant Interleukin- 4 (6ng/ml; R&D Systems) and IgE (100 ⁇ g/ml; Calbiochem). Cells are then maintained for 5 days at 37 0 C, 5% CO2 in a humidified atmosphere.
- Primed LAD2 cells are centrifuged (30Og, 5min), the supernatant discarded and the cell pellet re-suspended at 1x10 4 cells/ml in RPMI supplemented with glutamine (2mM). Following a further centrifugation (30Og, 5min) the cells are re-suspended in fresh RPMI with glutamine (2mM), adjusted to a density of 2.85x10 5 /ml, and pipetted into sterile V-well plates (70 ⁇ l_/well; Greiner) containing 20 ⁇ l diluted compound (prepared as detailed above).
- Cells are then incubated for 1h (37 0 C, 5% CO 2 in a humidified atmosphere) before activating with a sub-maximal concentration of anti- lgE (10 ⁇ l volume to give a final assay dilution of 1 :2700; Sigma).
- a sub-maximal concentration of anti- lgE 10 ⁇ l volume to give a final assay dilution of 1 :2700; Sigma.
- plates are centrifuged (120Og, 10 min, 4 0 C) and the supernatant removed for hexosaminidase assay.
- the cell pellet is lysed in 100 ⁇ l/well triton-X (0.5% in RPMI 2mM glutamine) at 37 0 C for 30 minutes.
- Beta-hexosaminidase activity is measured by the conversion of 4-methylumbelliferyl N-acetyl- ⁇ -D glucosaminide (Sigma) to a fluorescent product.
- a useful screening strategy comprises assay 1 (enzyme assay (pKi), assay 2 and then assay 3 (B Cell Proliferation) or assay 4 (LAD2). Intermediates and Examples
- DMSO dimethylsulfoxide
- DMF refers to ⁇ /, ⁇ /-dimethylformamide
- THF refers to tetrahydrofuran
- HPLC refers to high performance liquid chromatography.
- ADDP refers to 1 ,1'(azodicarbonyl)dipiperidine
- SPE refers to solid phase extraction cartridges marketed by lsolute
- TBTU refers to O-benzotriazol-1-yl- ⁇ /, ⁇ /, ⁇ /', ⁇ /',-bis(tetramethylene)uronium tetrafluoroborate
- HOBt refers to N-hydroxybenzotriazole hydrate
- “Hydrophobic frits” refers to filtration tubes sold by Whatman. SPE (solid phase extraction) refers to the use of cartridges sold by International Sorbent Technology Ltd.
- TLC thin layer chromatography
- LCMS was conducted on a Supelcosil LCABZ+PLUS column (3.3 cm x 4.6 mm ID) eluting with 0.1% HCO2H and 0.01 M ammonium acetate in water (solvent A) and 0.05% HCO2H 5% water in acetonitrile (solvent B), using the following elution gradient 0.0-7min 0%B, 0.7-4.2 min 100%B, 4.2-5.3 min 0%B, 5.3-5.5min 0%B at a flow rate of 3ml/min.
- the mass spectra were recorded on a Fisons VG Platform spectrometer using electrospray positive and negative mode (ES+ve and ES-ve).
- Flashmaster refers to an automated multi-user flash chromatography system, available from Argonaut Technologies Ltd, which utilises disposable, normal phase, SPE cartridges (2 g to 100 g). It provides quaternary on-line solvent mixing to enable gradient methods to be run. Samples are queued using the multi-functional open access software, which manages solvents, flow-rates, gradient profile and collection conditions.
- the system is equipped with a Knauer variable wavelength uv-detector and two Gilson FC204 fraction-collectors enabling automated peak cutting, collection and tracking.
- Preparative mass directed HPLC was conducted on a Waters FractionLynx system comprising of a Waters 600 pump with extended pump heads, Waters 2700 autosampler, Waters 996 diode array and Gilson 202 fraction collector on a 10 cm 2.54 cm ID ABZ+ column, eluting with either 0.1% formic acid or trifluoroacetic acid in water (solvent A) and 0.1% formic or trifluoroacetic acid in acetonitrile (solvent B) using the appropriate elution gradient.
- Mass spectra were recored on Micromass ZMD mass spectrometer using electrospray positive and negative mode, alternate scans. The software used was MassLynx 3.5 with OpenLynx and FractionLynx options.
- Ethyl chloroformate (136ml) was added dropwise to a chilled solution of 2,3-dihydro- 1,2-benzisothiazol-1 ,1 -dioxide (68.Og) in pyridine (340ml) over 1h, maintaining the temperature below 18 0 C.
- the slurry was stirred for 10min at 2O 0 C, diluted dropwise with water (1000ml) over 75min and stirred for a further 30min.
- the mixture was chilled to 0-5 0 C, aged for 1 h and isolated by filtration.
- the cake was washed with cold water (2x250ml), and dried in vacuo to give the title compound as a white solid (72.35g).
- a suspension of the above 6-nitro-2,3-dihydro-1 ,2-benzisothiazol-1,1 -dioxide (10.0g), 5%Pd/C (1.0g, 50%wet) and methanesulfonic acid (4.5ml) in propan-2-ol (100ml) was hydrogenated under hydrogen at 2bar pressure at 5O 0 C.
- the mixture was purged with nitrogen, cooled to 2O 0 C and treated with a mixture of 2M sodium hydroxide (90ml) and 10M sodium hydroxide (10ml).
- the reaction was stirred for 2h at ambient temperature and filtered through a filter pad, washing the pad with water (20ml).
- Zinc dust (2.4g) was added portion wise to a stirred suspension of 5-amino-1 ,2- benzisothiazol-3(2H)-one 1 ,1-dioxide (820mg) in concentrated hydrochloric acid (10ml). The mixture was stirred at room temperature for 20 hours before saturated aqueous sodium hydrogen carbonate solution was added to the mixture until the pH of the solution was 8. The mixture was filtered and extracted with ethyl acetate (4x150ml). The combined organic phases were dried (MgSO 4 ), filtered and the solvents evaporated in vacuo to give the title compound as a yellow solid (230mg). LC/MS: Rt 0.82min, MH + 185.
- Triethylamine (1.97g, 19.46mmol) was added to an ice-cooled solution of 2-amino-4- nitrophenol (3g, 19.46mmol) in THF (30ml).
- Bromodifluoroacetylbromide (2.48ml, 19.46mmol) was added dropwise to the solution and the mixture stirred at O 0 C for 45min.
- the reaction was quenched with water and extracted with ethyl acetate (3x 50ml).
- the crude product was purified by chromatography on a Flashmaster cartridge (Si, 7Og), eluting with a MeOH/DCM/triethylamine gradient (0-15% MeOH, 1% triethylamine) and then washing the column with further MeOH/DCM/triethylamine (10% MeOH, 1% triethylamine.
- the product fractions were reduced to dryness under vacuum to give 6-amino-2,2-difluoro-2H-1 ,4-benzoxazin-3(4H)-one (857mg) as a brown solid.
- LC/MS Rt 2.25min, [M-H]- 199.
- N,N,N'-diethylethylenediamine (Aldrich, 2.1g, 18.0mmol) was added to an ice-cooled solution of 3-nitrobenzenesulphonylchloride (2g, 9.0mmol) in DCM (15ml) and the reaction stirred at room temperature for 17hrs. Water (15ml) was added to the mixture, the phase separated (frit) and the organic phase applied directly to a biotage column (Si, 4Og).
- Iron powder (1.22g, 22.0mmol) was added in portions to an ice-cooled solution of N- [2-(diethylamino)ethyl]-3-nitrobenzenesulfonamide (1.66g, 5.5mmol) in acetic acid (60ml).
- the reaction was stirred at room temperature for 18hrs, diluted with water (250ml) and ethyl acetate (100ml) and solid sodium hydrogen carbonate added to the mixture until pH7 was achieved.
- the organic phase was separated, the aqueous extracted with ethyl acetate (3x 100ml).
- the combined organic phases were washed with water and brine, dried (magnesium sulphate) and reduced to dryness in vacuo.
- reaction mixture was was allowed to warm to room temperature overnight, then diluted with methylene chloride (100ml), washed with hydrochloric acid (1N, 3 x 150ml) and a solution of saturated sodium bicarbonate (3 x 150ml). The organic layer was dried over anhydrous sodium sulfate, gravity filtered, and concentrated under reduced pressure to afford 2-(3-nitrophenyl)- ⁇ /-[3-(2- oxo-1-pyrrolidinyl)propyl]acetamide as a yellow oil which was used, without purification, in the next reaction.
- Example 1 formic acid - 5- ⁇ [4-(1H-indazol-4-ylamino)-2-pyrimidiny ⁇ amino)-1 ,3- dihvdro-2H-indol-2-one (1 :1)
- Method A (default) - on a Supelcosil LCABZ+PLUS column (3um, 3.3cm x 4.6mm ID) eluting with 0.1% HCO 2 H and 0.01 M ammonium acetate in water (solvent A), and 95% acetonitrile and 0.05% HCO 2 H in water (solvent B), using the following elution gradient 0-0.7 minutes 0%B, 0.7-4.2 minutes 0 ⁇ 100%B, 4.2-5.3 minutes 100%B, 5.3-5.5 minutes 100 ⁇ 0%B, flow 3 ml/min.
- Mass spectra were recorded on a Fisons VG Platform mass spectrometer using electrospray positive ionisation [(ES+ve to give MH + and M(NH 4 ) 4" molecular ions] or electrospray negative ionisation [(ES-ve to give (M-H) " molecular ion] modes, or on a Perkin Elmer Sciex 100 atmospheric pressure ionization (APCI) mass spectrometer, or a Finnigan LCQ Duo LC/MS Ion Trap electrospray ionization (ESI) mass spectrometer. Retention time in LC/MS is referred to as "Rt" (time in minutes).
- Mass directed autoprep refers to methods where the material was purified by high performance liquid chromatography on a HPLCABZ+ 5 ⁇ m column (5cm x 10mm i.d.) with 0.1% HCO 2 H in water and 95% MeCN, 5% water (0.5% HCO 2 H) utilizing the following gradient elution conditions: 0-1.0 minutes 5%B, 1.0-8.0 minutes 5 ⁇ 30%B, 8.0-8.9 minutes 30%B, 8.9-9.0 minutes 30 ⁇ 95%B, 9.0-9.9 minutes 95%B, 9.9-10 minutes 95 ⁇ 0%B at a flow rate of 8ml minutes "1 (System 2).
- the Gilson 202-fraction collector was triggered by a VG Platform Mass Spectrometer on detecting the mass of interest.
- BiotageTM chromatography refers to purification carried out using equipment sold by Dyax Corporation (either the Flash 4Oi or Flash 15Oi) and cartridges pre-packed with KPSiI.
- Example 105 ⁇ /-(3-hvdroxypropyl)-2-IY4-(r4-(1 H-indazol-4-ylamino)-2- DvrimidinvliaminolphenvOoxvlacetamide:
- Example 106 Methyl ⁇ /- ⁇ [(4-(r4-(1H-indazol-4-ylamino)-2- pyrimidinyl1amino)phenyl)oxylacetyl) serinate
- the reaction mixture was stirred at room temperature for 15min before a solution of methyl serinate (Aldrich, 0.19g, 1.2mmol) and ⁇ /, ⁇ /-diisopropylethylamine (0.1Og, O. ⁇ mmol) in ⁇ /, ⁇ /-dimethylformamide (1.5ml) was added via syringe. After stirring for 15 h, the reaction mixture was diluted with ethyl acetate (100ml) and washed with water (3 x 50ml) and a 5% lithium chloride solution (50ml).
- Example 107 ⁇ /-(3-hvdroxypropyl)-1-(3-(r4-(1H-indazol-4-ylamino)-2- pyrimidinyl1amino ⁇ phenyl)methanesulfonamide
- reaction mixture was concentrated under reduced pressure, the residue diluted with methylene chloride (5ml), and trifluoroacetic acid (1 ml) was added. After stirring at room temperature for 30min, the reaction mixture was diluted with methylene chloride (50ml), and carefully neutralized by saturated sodium bicarbonate solution. The solid which had formed was collected by vacuum filtration, dissolved in methanol and the combined organic solvents were concentrated under reduced pressure.
- Example 108 ⁇ /-[3-(2,5-dioxo-1 -pyrrolidinyl)propy ⁇ -2-(4-(r4-(1 /-/-indazol-4-ylaminoV2- pyrimidinyllamino) phenvQacetamide
- reaction mixture was concentrated under reduced pressure, the residue diluted with methylene chloride (6ml), and trifluoroacetic acid (1ml) was added. After stirring at room temperature for 30min, the reaction mixture was agitated with a solution of saturated sodium bicarbonate (100ml).
- Example 109 2-(3-(f4-(1 /-/-indazol-4-ylamino)-2-pyrimidinyllamino)phenyl)-A/-r3-(2- oxo-1-pyrrolidinyl)propyllacetamide
- reaction mixture was concentrated under reduced pressure, the residue diluted with methylene chloride (6ml), and trifluoroacetic acid (1 ml) was added. After stirring at room temperature for 30min, the reaction mixture was agitated with a solution of saturated sodium bicarbonate (100ml).
- Example 110 1 ,1 -dimethylethylf(4- ⁇ f4-(1 H-indazol-4-ylamino)-2-pyrimidinv ⁇ amino) phenyQoxylacetate
- Example 111 1-(4- ⁇ r4-(1H-indazol-4-ylamino)-2-pyrimidinyllamino)phenvn-/V- methylmethanesulfonamide
- Example 114 r(4- ⁇ f4-(1/-/-indazol-4-ylamino)-2-pyrimidinyllamino)phenyl)oxylacetic acid hydrochloride
- Example 116 ⁇ /-(3,4-dimethyl-5-isoxazolyl)-1-(4- ⁇ f4-(1H-indazol-4-ylamino)-2- pyrimidinyllamino)phenyl)methanesulfonamide
- Example 117 ⁇ /-(4,5-dimethyl-1.3-thiazol-2-ylV1-f4-ff4-(1 Wndazol-4-ylamino)-2- pyrimidinvnamino ⁇ phenyl)methanesulfonamide
- Example 118 1-(4- ⁇ f4-(1f/-indazol-4-ylamino)-2-pyrimidinyllamino)phenyl)-A/-f3-(2- oxo-1-pyrrolidinyl)propyllmethanesulfonamide
- Example 119 ⁇ /-(3-hvdroxypropyl)-1-(4- ⁇ f4-(1/-/-indazol-4-ylamino)-2- pyrimidinyllamino ⁇ phenyl)methanesulfonamide
- Example 120 2-[(4-([4-(I /-/-indazol-4-ylamino)-2-pyrimidinyllamino ⁇ phenyl)oxy1-A/-r3- (2-oxo-1-pyrrolidinyl)propyllacetamide
- Example 121 ⁇ /-(3,4-dimethyl-5-isoxazolyl)-2-(4-ff4-(1 A7-indazol-4-ylamino)-2- pyrimidinv ⁇ aminolphenvPacetamide
- N-(2-chloro-4-pyrimidinyl)-1 H-indazol-4-amine (280mg, 1.1mmol) and 2-(4- aminophenyl)-/v-(3,4-dimethyl-5-isoxazolyl)acetamide (280mg, 1.1 mmol) were dissolved in IPA (7 ml) with a few drops of 2N HCI in ether. The reaction was heated for 3 days and then concentrated in vacuo. The mixture was dissolved in methylene chloride (5ml) treated with TFA (3ml) and stirred at room temperature for 1h and then quenched with NaHC ⁇ 3. The resulting solid was filtered and purified by flash chromatography to yield the title compound.
- Example 123 N- ⁇ 4-r(difluoromethv0oxylphenyl)-2-(4- ⁇ l4-(1 /-/-indazol-4-ylamino)-2- Pyrimidinyllamino)phenvOaceta ⁇ iide
- Example 124 ⁇ /M H-indazol-4-yl- ⁇ / 2 -(4-r(methylsulfonyl)methyllphenyl>-2,4- pyrimidinediamine
- Example 125 2-(4-(K-(I H-indazol-4-ylamino)-2-pyrimidinyllamino
- Example 127 4- ⁇ r4-(1/-/-indazol-4-ylaminoV2-pyrimidinyl1amino)benzoic acid
- Example 129 2-(4-(F4-(1 f/-indazol-4-ylamino)-2-pyrimidinvnamino ⁇ phenyl)- ⁇ /-f4- (trifluoromethyl)phenyllacetamide
- Example 131 ⁇ /-(3-chlorophenvO-2-(4-(r4-(1 /-/-indazol-4-ylamino)-2- pyrimidinyliaminolphenyPacetamide
- Example 132 ⁇ /-(4-chlorophenvP-2-(4-(r4-(1 H-indazol-4-ylamino)-2- pyrimidinvnamino)phenyl)acetamide
- Example 133 ⁇ /-(4,5-dimethyl-1.3-thiazol-2-ylV2-(4- ⁇ f4-(1H-indazol-4-ylamino)-2- pyrimidinv ⁇ amino ⁇ phenyl)acetamide
- Example 134 2-(4- ⁇ f4-f1 H-indazol-4-ylamino)-2-pyrimidinyllamino)phenyl)- ⁇ /-r3-(2- oxo-1-pvrrolidinyl)propvllacetamide
- Example 136 ⁇ /-(4-(r4-(1/-/-indazol-4-ylamino)-2- pyrimidiny ⁇ amino)phenyl)methanesulfonamide
- Example 138 2-(4- ⁇ r4-(1H-indazol-4-ylamino)-2-pyrimidinyl1amino)phenyl)- ⁇ /-3- pyridinylacetamide
- Example 140 ⁇ /M H-indazol-4-yl- ⁇ / 2 -r4-(1 H-tetrazol-5-ylmethvnphenvn-2.4- pyrimidinediamine
Abstract
Bis-anilinopyrimidine compounds of the formula (I); are useful as inhibitors of spleen tyrosine kinase (Syk) and thus useful in treating diseases resulting from in appropriate mast cell activation, for instance allergic and inflammatory diseases.
Description
Novel compounds
The present invention relates to novel bis-anilinopyrimidine compounds which have activity against the spleen tyrosine kinase (Syk kinase), processes for their preparation, pharmaceutically acceptable formulations containing them and their use in therapy.
Allergic rhinitis and asthma are diseases associated with hypersensitivity reactions and inflammatory events involving a multitude of cell types including mast cells, eosinophils, T cells and dendritic cells. Following exposure to allergen, high affinity immunoglobulin receptors for IgE (FcεRl) and IgG (FcγRI) become cross-linked and activate downstream processes in mast cells and other cell types leading to the release of pro-inflammatory mediators and airway spasmogens. In the mast cell, for example, IgE receptor cross-linking by allergen leads to release of mediators including histamine from pre-formed granules, as well as the synthesis and release of newly synthesised lipid mediators including prostaglandins and leukotrienes.
Syk kinase is a non-receptor linked tyrosine kinase which is important in transducing the downstream cellular signals associated with cross-linking FcεRl and or FcγRI receptors, and is positioned early in the signalling cascade. In mast cells, for example, the early sequence of FcεRl signalling following allergen cross-linking of receptor-lgE complexes involves first Lyn (a Src family tyrosine kinase) and then Syk kinase. Inhibitors of Syk kinase activity would therefore be expected to inhibit all downstream signalling cascades thereby alleviating the immediate allergic response and adverse events initiated by the release of pro-inflammatory mediators and spasmogens (Wong, B., Grossbard, E. B. Payan, D. G & Masuda, E. S. Expert Opin. Investig. Drugs (2004) 13 (7) 743-762).
Recently, it has been shown that the Syk kinase inhibitor R112 (Rigel), dosed intranasal^ in a phase I/I I study for the treatment of allergic rhinitis, gave a statistically significant decrease in PGD2, a key immune mediator that is highly correlated with improvements in allergic rhinorrhea, as well as being safe across arrange of indicators, thus providing the first evidence for the clinical safety and efficacy of a topical Syk kinase inhibitor. (Guyer B, Shimamoto S, Bradhurst A et al, J Allergy Clin Immunol., (2004) 113(2):S28-29). In a more recent phase Il clinical trial for allergic rhinitis (Clinical Trials.gov Identifier NCT0015089), R112 was however shown as having a lack of efficacy versus placebo.
Rheumatoid Arthritis (RA) is an auto-immune disease affecting approximately 1% of the population. It is characterised by inflammation of articular joints leading to debilitating destruction of bone and cartilage. Recent clinical studies with Rituximab,
which causes a reversible B cell depletion, (J. CW. Edwards et al 2004, New Eng. J. Med. 350: 2572-2581) have shown that targeting B cell function is an appropriate therapeutic strategy in auto-immune diseases such as RA. Clinical benefit correlates with a reduction in auto-reactive antibodies (or Rheumatoid Factor) and these studies suggest that B cell function and indeed auto-antibody production are central to the ongoing pathology in the disease.
Studies using cells from mice deficient in the Syk kinase have demonstrated a non- redundant role of this kinase in B cell function. The deficiency in Syk kinase is characterised by a block in B cell development (M. Turner et al 1995 Nature 379: 298-302 and Cheng et al 1995, Nature 378: 303-306). These studies, along with studies on mature B cells deficient in Syk kinase (Kurasaki et al 2000, Immunol. Rev. 176:19-29), demonstrate that Syk kinase is required for the differentiation and activation of B cells. Hence, inhibition of Syk kinase in RA patients, is likely block B cell function and hence to reduce Rheumatoid Factor production. In addition to the role of Syk kinase in B cell function, of relevance to the treatment of RA, is the requirement for Syk kinase activity in Fc receptor (FcR) signalling. FcR activation by immune commplexes in RA has been suggested to contribute to the release of multiple pro-inflammatory mediators.
The contribution of Syk kinase dependent processes to the pathology of RA has been reviewed in Wong et al (2004, ibid).
WO 03/057695 (Boehringer lngelheim Pharmaceuticals, Inc.) describes substituted [1 ,6]-naphthyridines that inhibit Syk kinase.
WO2003/063794, WO2004/014382, WO2005/012294 and WO2005/16893 (Rigel Pharmaceuticals, Inc) describes a series of 2,4-pyrimidinediamaine compounds which inhibit Syk kinase, for use in treating autoimmune diseases.
WO2005/026158 (Novartis AG) describes 2,4-di (hetero)-arylamino-pyrimidine derivatives which have ZAP-70 and/or Syk inhibitory activities.
WO 04/035604 discloses the structural co-ordinates of the human Syk protein.
There remains however the need to identify further compounds which are inhibitors of Syk kinase.
Thus, in a first aspect invention, the present invention provides a compound of formula (I):

in which:
R1, R2 and R3 is each independently selected from hydrogen, halogen, hydroxy, -C1-6 alkyl, -NR5R6, -C1-6 alkylene-NR5R6, -CN, -C^alkylene-COaH, C(O)Ci-6 alkoxy, -C1-2 alkyl substituted by 1 or more fluorine atoms), -C1-6 alkoxy, -C(O)C1-6alkyl, - C(O)NR5R6, -C(O)NHR7, -OCH2C(O)NHR8, -OCH2C(O)OR5, -CH2C(O)NHR8, -NR5C(O)R6, -SC1-6alkyl, -S(O)C1-6alkyl, -S(O)2C1-6alkyl, -CH2S(O)2C1-6alkyl, - NHS(O)2R9, -S(O)3H, -S(O)2NR5R6, -S(O)2NR5R10, or -CH2S(O)2NR5R10, such that at least one of R1, R2 and R3 is hydrogen; or
R1 and R3 is each hydrogen and R2 is C3.7cycloalkyl, benzyloxy, benzimidazol-2yl, a 5- or 6-membered heteroaryl ring or a heterocyclic ring comprising from 1 to 3 heteroatoms selected from O, N and S bonded to the phenyl ring through a ring carbon or nitrogen (if present) atom and which heterocyclic ring is unsubstituted or substituted on one or more ring carbon atoms by C1-6alkyl or oxo (=0), or on a ring nitrogen atom by Ci.6alkyl; or
R1 is hydrogen, halogen or C1-6 alkyl and R2 and R3 are attached to adjacent carbon atoms of the phenyl ring and are joined and together to form, in combination with the carbon atoms on the phenyl group to which they are attached, a 5- or 6-membered heteroaryl or heterocyclic ring ; or
R1 is hydrogen, halogen or C1-6 alkyl and R2 and R3 are attached to adjacent carbon atoms of the phenyl ring and are joined together to form, in combination with the carbon atoms on the phenyl group to which they are attached, a 5- or 6-membered carbocyclic ring, and
R5 and R6 are is each independently hydrogen or C1-6alkyl, or R5 and R6 together with the nitrogen atom to which they are attached form a 5- or 6-membered heterocyclic ring which may comprise a second heteroatom selected from O, S or N and which may be substituted on the second N, if present, by C1-6alkyl , and which may be substituted on a ring carbon by oxo (=0), C1-6alkyl, di- C1-6alkyl (which may be the same or different), or halogen;
R7 is phenyl optionally substituted by halogen, C1-6alkyl;
R8 is hydrogen, C1-6alkyl optionally terminally substituted with hydroxy, 2-oxo-1- pyrrolidinyl or 2,5-dioxo-1-pyrrolidinyl, a 5- or 6-membered heteroaryl ring comprising
1 or 2 heteroatoms independently selected from O, N and S and optionally substituted on a ring carbon by C1-6alkyl, phenyl optionally substituted by halo, Crealkyl. C1-6alkoxy or trifluoromethyl, or, together with the nitrogen to which it is attached is a C^alkyl ester of serine;
R9 is Ci-6 alkyl or phenyl optionally substituted by C-,.6alkyl;
R10 is Ci-6alkoxy, Ci-6alkyl optionally terminally substituted with hydroxyl, 2-oxo-1- pyrrolidinyl or 2,5-dioxo-1-pyrrolidinyl, or R10 is a 5 or 6-membered heteroaryl ring comprising 1 heteroatom selected from O, N and S or 2 different heteroatoms independently selected from O, N and S and optionally substituted on one or more ring carbons by C1-6alkyl;
a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
In a second aspect, the present invention provides for a compound of formula (I) which is of the formula (Ia):

in which:
R1, R2a and R3a is each independently selected from the group of substituents Group A which comprises hydrogen, halogen, hydroxy, -C1-6 alkyl, -NR5R6, -Ci.6 alkylene-NR5R6, -CN, -Co-3alkylene-C02H, C(O)C1-6 alkoxy, -Ci-2 alkyl substituted by 1 or more fluorine atoms, -C1-6 alkoxy, -CCOJCLβalkyl, -C(O)NR5R6, -C(O)NHR7, -OCH2C(O)NHR8, -OCH2C(O)OR5, -CH2C(O)NHR8, -NR5C(O)R6, -SC1- βalkyl, -S(O)C1-6alkyl, -S(O)2C1-6alkyl, -CH2S(O)2C1-6alkyl, -NHS(O)2R9, -S(O)3H, -S(O)2NR5R6, -S(O)2NR5R10, or -CH2S(O)2NR5R10, such that at least one of R1, R2 and R3 is hydrogen; or
R1 is hydrogen and one of R2a and R3a is hydrogen and the other is R4 wherein R4 is benzyloxy, benzimidazol-2yl, a 5- or 6-membered heteroaryl ring or a heterocyclic ring comprising from 1 to 3 heteroatoms selected from O, N and S bonded to the phenyl ring through a ring carbon or nitrogen (if present) atom and which heterocyclic ring is unsubstituted or substituted on one or more ring carbon atoms by Ci-6alkyl or oxo (=0), or on a ring nitrogen atom by Ci-6alkyl; or
R1 is hydrogen, halogen or C1-6 alkyl and R2a and R3a are joined and together form, in combination with the carbon atoms on the phenyl group to which they are attached, a 5- or 6-membered ring wherein: the ring is heteroaryl or heterocyclic and comprises one heteroatom selected from O, N or S, the ring is heteroaryl or heterocyclic and comprises two heteroatoms selected from O, N or S, which maybe the same or different, and excluding two S atoms, the ring is heteroaryl and comprises three heteroatoms of which two are N atoms and the third is nitrogen or oxygen, which ring may be unsubstituted or substituted on: one or more ring carbon atoms which each may be independently substituted by oxo (=0) or by 1 or 2 substituents which may be the same or different selected from C1-6 alkyl, hydroxyl, or halo, one or more ring nitrogen atoms by C1-6 alkyl or C1-6alkylcarbonyl, and a ring sulphur atom by (O)2; or
R1 is hydrogen, halogen or C1-6 alkyl and R2a and R3a are joined and together form, in combination with the carbon atoms on the phenyl group to which they are attached, a 5- or 6- membered non-aromatic carbocyclic ring which may be unsubstituted or substituted by oxo (=0), and
R5 and R6 are is each independently hydrogen or C1-6alkyl;
R7, R8, R9 and R10 are as hereinbefore defined for formula (I); or
a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
Compounds of the present invention are useful as inhibitors of Syk and thus useful in treating diseases resulting from in appropriate mast cell activation, for instance allergic and inflammatory diseases.
In a further embodiment, two of R1, R2 and R3 are hydrogen.
In a further embodiment, representative values for substituents in Group A include: hydroxy, chloro, methyl, (1-methylethyl), -CONH2, -CONHCH3, -CONH(C2H5), -CON(C2Hs)2, -CN, -CF3, methyloxy, (i-methylethyl)oxy, -CO2H, -CH2CO2H, - C(O)OCH3,-SO2NH2, and -SO2CH3.
In a further embodiment, R1 and R3 is each hydrogen and R2 is R4.
In a further embodiment, R1 and R2 is each hydrogen and R3 is R4.
In a further embodiment, representative examples of R4 include: benzimidazol-2-yl, tetrazol-5-yl ;

In a further embodiment, representative examples of the 5- or 6- membered saturated or unsaturated ring formed by R2a and R3a, fused with the phenyl ring, include:


In a further embodiment, representative examples of the 5- or 6- membered saturated or unsaturated ring formed by R2a and R3a, fused with the phenyl ring, and including any substituents which may be present, include:



In a further embodiment, representative examples of the 5- or 6- membered saturated carbocyclic ring formed by R2a and R3a include:

Representative examples, including substituents, include:

In a further embodiment, representative examples of R5 and R6 include: H, methyl, and ethyl.
In a further embodiment, representative examples of R7 include phenyl and 4- methyl phenyl.
In a further embodiment, representative examples of R8 include: hydrogen, 3- hydroxypropyl, 2,5-dioxo-1-pyrrolidinyl, 2-oxo-1-pyrrolidinyl, difluoromethyloxy, methyl serinate (including N);

Representative examples of compounds of formula (I) include:
6-{[4-(1 H-indazol-4-ylamino)-2-pyrimidinyl]amino}-3,4-dihydro-1 (2H)-naphthalenone;
N2-[3,4-bis(methyloxy)phenyl]-N4-1H-indazol-4-yl-2,4-pyrimidinediamine;
5-{[4-(1 H-indazol-4-ylamino)-2-pyrimidinyl]amino}-1 ,3-dimethyl-1 ,3-dihydro-2H- benzimidazol-2-one;
N-(3-{[4-(1 H-indazol-4-ylamino)-2-pyrimidinyl]amino}phenyl)methanesulfonamide;
N2-[3,5-bis(methyloxy)phenyl]-N4-1H-indazol-4-yl-2,4-pyrimidinediamine;
N4-1H-indazol-4-yl-N2-(1-methyl-1 H-benzimidazol-5-yl)-2,4-pyrimidinediamine;
N4-1 H-indazol-4-yl-N2-(1 -methyl-1 H-indazol-5-yl)-2,4-pyrimidinediamine;
N4-1 H-indazol-4-yl-N2-[3-(methylsulfonyl)phenyl]-2,4-pyrimidinediamine;
N2-[3-chloro-4-(methyloxy)phenyl]-N4-1H-indazol-4-yl-2,4-pyrimidinediamine;
2-[(3-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyl]amino}phenyl)oxy]-N-methylacetamide;
6-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyl]amino}-2-benzofuran-1(3H)-one;
N4-1H-indazol-4-yl-N2-[3-(1 ,3-oxazol-5-yl)phenyl]-2,4-pyrimidinediamine;
N4-1H-indazol-4-yl-N2-[3-(5-methyl-1 ,2,4-oxadiazol-3-yl)phenyl]-2,4- pyrimidinediamine;
N-ethyl-4-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyl]amino}benzamide;
N2-1 ,3-benzodioxol-5-yl-N4-1 H-indazol-4-yl-2,4-pyrimidinediamine; N4-1 H-indazol-4- yl-N2-(1 -methyl-1 H-indazol-6-yl)-2,4-pyrimidinediamine;
N4-1 H-indazol-4-yl-N2-[3-(1 H-pyrazol-3-yl)phenyl]-2,4-pyrimidinediamine;
N4-1 H-indazol-4-yl-N2-1 H-indazol-5-yl-2,4-pyrimidinediamine;
N4-1 H-indazol-4-yl-N2-[3-(methyloxy)phenyl]-2,4-pyrimidinediamine;
3-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyl]amino}benzonitrile;
N4-1 H-indazol-4-yl-N2-[3-(trifluoromethyl)phenyl]-2,4-pyrimidinediamine;
N4-1 H-indazol-4-yl-N2-[3-(4-methyl-1 ,3-oxazol-5-yl)phenyl]-2,4-pyrimidinediamine;
3-{[4-(1 H-indazol-4-ylamino)-2-pyrimidinyl]amino}phenol; and methyl 3-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyl]amino}benzoate;
or salt or solvate thereof, in particular, a pharmaceutically acceptable salt or solvate thereof.
It will be understood that compounds of formula (I) and formula (Ia) may exist in alternative tautomeric forms, for example when R2a represents a non-hydrogen substituent on the nitrogen atom in the 2-position.
When used herein, and unless otherwise indicated, the term "heteroaryl" includes single or fused rings comprising up to four hetero-atoms in the ring selected from
oxygen, nitrogen and sulphur and optionally substituted with up to three substituents. Representative heteroaryl rings comprise from 4 to 7, preferably 5 or 6, ring atoms. A fused heteroaryl ring system may include carbocyclic rings and need only include one heterocyclic ring.
When used herein, and unless otherwise indicated, the term "heterocyclic" includes non-aromatic single or fused rings comprising up to four hetero-atoms in the ring selected from oxygen, nitrogen and sulphur and optionally substituted with up to three substituents. Representative heterocyclic rings comprise from 4 to 7, preferably 5 to 6, ring atoms. A fused heterocyclic ring system may include carbocyclic rings and need only include one heterocyclic ring.
Examples of substituents for such heteroaryl and heterocyclic rings include Ci-6 alkyl, halogen, C1-6 alkoxy, cyano, hydroxy, nitro, amino, -N(CH3)2, -NHC(O)Ci-6 alkyl, - CF3, -C(O)OCi-6 alkyl, -C(O)NR19R20 (wherein R19 and R20 independently represent hydrogen or C1-6 alkyl, e.g., methyl), -S(O)2N(CHs)2, or -S(O)2CH3, and, for heterocyclic, oxo (=0).
Examples of "-C1-2 alkyl substituted by 1 or more fluorine atoms" include, but is not restricted to, -CF3.
References to alkyl include references to both straight chain and branched chain aliphatic isomers of the corresponding alkyl. It will be appreciated that references to alkylene and alkoxy shall be interpreted similarly.
References to C3-7 cycloalkyl include references to all alicyclic (including branched) isomers of the corresponding alkyl.
When used herein, and unless otherwise indicated, the term "carbocyclic" refers to rings which may be saturated or unsaturated but which are not aromatic, may be branched, which contain 5-7 carbon atoms and which may be optionally substituted with up to three substituents.
Examples of substituents for such carbocyclic rings include those previously mentioned for heteroaryl and heterocyclic.
When used herein, the term "pharmaceutically acceptable" refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio. The skilled artisan will appreciate that pharmaceutically acceptable salts of the compound of the present invention may be prepared. As used herein, the term "pharmaceutically acceptable
salts" refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects. These pharmaceutically acceptable salts may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively. Indeed, in certain embodiments of the invention, pharmaceutically acceptable salts may be preferred over the respective free base or free acid because such salts impart greater stability or solubility to the molecule thereby facilitating formulation into a dosage form.
In certain embodiments, the compound of the present invention may contain one or more acidic functional groups. Suitable pharmaceutically acceptable salts include salts of such acidic functional groups. Representative salts include pharmaceutically acceptable metal salts such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc salts; carbonates and bicarbonates of a pharmaceutically- acceptable metal cation such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc; pharmaceutically acceptable organic primary, secondary, and tertiary amines including aliphatic amines, aromatic amines, aliphatic diamines, and hydroxy alkylamines such as methylamine, ethylamine, 2-hydroxyethylamine, diethylamine, triethylamine, ethylenediamine, ethanolamine, diethanolamine, and cyclohexylamine.
Compounds of the present invention are basic and accordingly generally capable of forming pharmaceutically acceptable acid addition salts by treatment with a suitable acid. Suitable acids include pharmaceutically acceptable inorganic acids and pharmaceutically acceptable organic acids. Representative pharmaceutically acceptable acid addition salts include hydrochloride, hydrobromide, nitrate, methylnitrate, sulfate, bisulfate, sulfamate, phosphate^, acetate, hydroxyacetate, phenylacetate, propionate, butyrate, isobutyrate, valerate, maleate, hydroxymaleate, acrylate, fumarate, malate, tartrate, citrate, salicylate, p-aminosalicyclate, glycollate, lactate, heptanoate, phthalate, oxalate, succinate, benzoate, o-acetoxybenzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, mandelate, tannate, formate, stearate, ascorbate, palmitate, oleate, pyruvate, pamoate, malonate, laurate, glutarate, glutamate, estolate, methanesulfonate (mesylate), ethanesulfonate (esylate), 2-hydroxyethanesulfonate, benzenesulfonate (besylate), p-aminobenzenesulfonate, p-toluenesulfonate (tosylate), and napthalene-2-sulfonate.
As used herein, the term "compound" refers to one or more compounds. Similarly, the term "a compound of the present invention" refers to one or more compounds of the present invention.
The compound of the present invention may exist in solid or liquid form. In the solid state, the compound of the present invention may exist in crystalline or noncrystalline form, or as a mixture thereof. For a compound of the present invention that is in crystalline form, the skilled artisan will appreciate that pharmaceutically acceptable solvates may be formed wherein solvent molecules are incorporated into the crystalline lattice during crystallization. Solvates may involve non-aqueous solvents such as, but not limited to, ethanol, isopropanol, DMSO, acetic acid, ethanolamine, and ethyl acetate, or they may involve water as the solvent that is incorporated into the crystalline lattice. Solvates wherein water is the solvent incorporated into the crystalline lattice are typically referred to as "hydrates." Hydrates include stoichiometric hydrates as well as compositions containing variable amounts of water. The invention includes all such solvates.
The skilled artisan will further appreciate that certain compounds of the invention that exist in crystalline form, including the various solvates thereof, may exhibit polymorphism (i.e. the capacity to occur in different crystalline structures). These different crystalline forms are typically known as "polymorphs." The invention includes all such polymorphs. Polymorphs have the same chemical composition but differ in packing, geometrical arangement, and other descriptive properties of the crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deformability, stability, and dissolution properties. Polymorphs typically exhibit different melting points, IR spectra, and X-ray powder diffraction patterns, which may be used for identification. The skilled artisan will appreciate that different polymorphs may be produced, for example, by changing or adjusting the reaction conditions or reagents, used in making the compound. For example, changes in temperature, pressure, or solvent may result in polymophs. In addition, one polymorph may spontaneously convert to another polymorph under certain conditions.
The compound of the present invention may contain one or more asymmetric centers (also referred to as a chiral center) and may, therefore, exist as individual enantiomers, diastereoisomers, or other stereoisomeric forms, or as mixtures thereof. Chiral centers, such as chiral carbon atoms, may also be present in a substituent such as an alkyl group. Where the stereochemistry of a chiral center is not specified the structure is intended to encompass any stereoisomer and all mixtures thereof. Thus, the compound of the present invention containing one or more chiral centers may be used as racemic mixtures, enantiomerically enriched mixtures, or as enantiomerically pure individual stereoisomers. Generally it is preferred to use a compound of formula (I) in the form of a purified single enantiomer.
Individual stereoisomers of a compound according to Formula (I) which contain one or more asymmetric center may be resolved by methods known to those skilled in the
art. For example, such resolution may be carried out (1) by formation of diastereoisomeric salts, complexes or other derivatives; (2) by selective reaction with a stereoisomer-specific reagent, for example by enzymatic oxidation or reduction; or (3) by gas-liquid or liquid chromatography in a chiral enviornment, for example, on a chiral support such as silica with a bound chiral ligand or in the presence of a chiral solvent. The skilled artisan will appreciate that where the desired stereoisomer is converted into another chemical entity by one of the separation procedures described above, a further step is required to liberate the desired form. Alternatively, specific stereoisomers may be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
The compound of the present invention may also contain double bonds or other centers of geometric asymmetry. Where the stereochemistry of a centre of geometric asymmetry is not specified, the structure is intended to encompass the trans (E) geometric isomer, the cis (Z) geometric isomer, and all mixtures thereof. Likewise, all tautomeric forms are also included the compound of the present invention whether such tautomers exist in equilibrium or predominately in one form.
Compounds of the present invention, as well as salts and solvates thereof, may be prepared by one or more of the general synthetic schemes described hereinafter.
Scheme 1

(see Bioorg + Med Chem Lett 11 (17) 2235-39 2001)
Scheme 2A

Scheme 2B

Scheme 2C
t-butyl nitrite


(I)
in which schemes R1, R2, and R3 are as hereinbefore defined, L1 and L2 are leaving groups and SEM is 2-(trimethylsilyl)ethoxymethyl.
Accordingly, in a further aspect, the present invention provides a process for preparing a compound of formula (I), or a salt or solvate thereof, which process comprises:
(a) reacting a compound of formula (II):

or a protected derivative thereof, wherein R1, R2, and R3 are as defined above and L1 represents a suitable leaving group, with a compound of formula (III)

or a protected derivative thereof; or
(b) reacting a compound of formula (IV):

or a protected derivative thereof wherein L2 represents a suitable leaving group, with a compound of formula (V)

or a protected derivative thereof wherein R , R and R are as defined above.
Process (a) may be performed in the presence of a solvent (for example, aqueous acetone) and optionally in the presence of dilute acid such as hydrochloric acid at a suitable temperature, preferably in the range of 0-100° C. Examples of suitable leaving groups (L1) include, but are not restricted to, an alky! and aryl sulfide, an alkyl and aryl sulfinyl, an alkyl and aryl sulfonyl, a halide (such as chloride, bromide or iodide), and an alcohol derived leaving group (such as triflate, mesylate, methylsulfonate or tosylate).
Alternatively process (a) may be performed in the presence of a non-nucleophilic base such as triethylamine or diisopropylethylamine, as a melt, at a suitable temperature, preferably in the range of 0-180° C.
Process (b) may be performed under conditions analogous to those described for process (a) above. Suitable leaving groups for L2 are also described above for L1.
Examples of protecting groups and the means for their removal can be found in T. W. Greene 'Protective Groups in Organic Synthesis' (J. Wiley and Sons, 1991). Suitable amine protecting groups include, but are not restricted to, sulphonyl (such as tosyl), acyl (such as benzyloxycarbonyl or t-butoxycarbonyl) and arylalkyl (such as benzyl), which may be removed by hydrolysis or hydrogenolysis as appropriate. Other suitable amine protecting groups include trifluoroacetyl (-C(O)CF3), which may be removed by base catalysed hydrolysis, or a solid phase resin bound benzyl group, such as a Merrifield resin bound 2,6-dimethoxybenzyl group (Ellman linker) which may be removed by acid catalysed hydrolysis (using, for example, trifluoroacetic acid).
Compounds of formula (II) may be prepared according to a process comprising: (a) reacting a compound of formula (Vl):

or a protected derivative thereof wherein R1, R2, and R3 are as defined above, with a suitable halogenating agent, such as POCI3, POBr3 or PCI5. This halogenation reaction is advantageously carried out at a temperature in the range of 50-150°C. If necessary, the halogen atom may be converted to another leaving group by conventional means; or
(b) reacting a compound of formula (VII):

wherein L1 and L3 represent leaving groups, with a compound of formula (V) as defined above.
Process (b) may be performed under analogous conditions as those described above for part (a). The product of this particular process is advantageously purified by methods well known to persons skilled in the art, such as flash chromatography or high performance liquid chromatography. Suitable leaving groups L3 include those mentioned above for L1, especially halide.
Compounds of formula (III) may be prepared by reducing a corresponding compound of formula (VIlI):

Reduction may be performed using reagents such as, but not limited to, hydrogen gas over palladium, iron metal with hydrochloric acid, stannous chloride and hydrochloric acid, or sodium dithionite, as appropriate under conventional conditions.
Compounds of formula (VIII) may be prepared by reacting an organic nitrite derivative, such as tert-butyl nitrite, with a compound of formula (IX):

Certain compounds of formula (IV) may be prepared by a process comprising reacting a compound of formula (VII) or a protected derivative thereof, wherein L1 and L3 represent suitable leaving groups, with a compound of formula (111) or a protected derivative thereof. This process may be performed under basic conditions,
such as tert-butanol and potassium carbonate, and advantageously at a temperature in the range of 50 to 1500C.
Compounds of formula (IV) may also be prepared from an analogous compound where the group L2 is a hydroxyl group (which can be converted to a halogen by reaction with POCI3 or PCI5) or a methylthio group (which can be converted to a methylsulfinyl or methylsulfonyl group by oxidation with metachloroperoxybenzoic acid or ozone, for example). Compounds in which L2 is a hydroxyl group may be prepared from corresponding compounds in which L is a methylthio group by oxidation with hydrogen peroxide in acetic acid.
Compounds of formula (IV) may also be prepared by reacting a compound of formula

The aforementioned reaction may be performed under nitrogen atom protecting conditions (for example, in the presence of acetate) to yield a product in which an indazolyl nitrogen atom is protected.
Compounds of formula (X) may be prepared by reducing the corresponding nitro compound using conventional conditions, such as sodium dithionite, hydrogen gas in the presence of palladium, or iron metal with hydrochloric acid. Such corresponding nitro compounds may be prepared by reacting a compound of formula (VII) with a compound of formula (Xl):

Compounds of formula (II), (IV), (VII), and (X), in which the leaving group represents other than halogen, may be obtained via the corresponding halogen by conventional methods.
Compounds of formula (V), (VII), (IX), and (Xl) are either known or may be prepared in accordance with known procedures.
Certain compounds of formula (II), (III), (IV), and (V) are new and form a further aspect of the invention.
Compounds of the present invention are useful as inhibitors of Syk and thus useful in treating diseases resulting from inappropriate mast cell activation, for instance allergic and inflammatory diseases.
Thus, in a further aspect, the present invention provides for a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, for use in therapy.
In a further embodiment, the present invention provides for a method of treating inappropriate mast cell activation which method comprises administering to a patient in need thereof an effective compound of formula I, or a or a pharmaceutically acceptable salt or solvate thereof.
In a further aspect, the present invention provides a method comprising administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, to inhibit a Syk kinase.
In a further aspect, the present invention provides a method of treating an inflammatory disease which comprises administering to a patient in need thereof an effective compound of formula I, or a pharmaceutically acceptable salt or solvate thereof.
In a further aspect, the present invention provides a method of treating an allergic disorder which comprises administering to a patient in need thereof an effective compound of formula I, or a pharmaceutically acceptable salt or solvate thereof.
Diseases and pathological conditions thought to be mediated by Syk kinase include inflammatory and allergic disorders involving mast cell activation, such as chronic obstructive pulmonary disease (COPD), adult respiratory distress syndrome (ARDS), asthma, ulcerative colitis, Crohn's Disease, bronchitis, conjunctivitis, psoriasis, sclerodoma, urticaria, dermatitis, and allergic rhinitis. They also include inflammatory conditions which involve B cells, for instance lupus and rheumatoid arthritis.
Likely disease targets for compounds of the present invention include chronic obstructive pulmonary disease (COPD), asthma and allergic rhinitis.
In a further aspect, the present invention provides a method of treating chronic obstructive pulmonary disease which comprises administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof.
In a further aspect, the present invention provides a method of treating asthma which comprises administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof.
In a further aspect, the present invention provides a method of treating allergic rhinitis which comprises administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof.
Compounds of the present invention may also be used in combination with other therapeutic agents, for instance an anti-inflammatory steroid, in particular an antiinflammatory corticosteroid.
Anti-inflammatory corticosteroids are well known in the art. Representative examples include fluticasone propionate (e.g. see US patent 4,335,121), beclomethasone 17- propionate ester, beclomethasone 17,21-dipropionate ester, dexamethasone or an ester thereof, mometasone or an ester thereof (e.g. mometasone furoate), ciclesonide, budesonide, and flunisolide. Further examples of anti-inflammatory corticosteroids are described in WO 02/12266 A1 (Glaxo Group Ltd), in particular, the compounds of Example 1 ( 6α,9α-difluoro-17α-[(2-furanylcarbonyl)oxy]-11β-hydroxy- 16α-methyl-3-oxo-androsta-1,4-diene-17β-carbothioic acid S-fluoromethyl ester) and Example 41 (6α,9α-difluoro-11 β-hydroxy-16α-methyl-17α-[(4-methyl-1 ,3-thiazole-5- carbonyl)oxy]-3-oxo-androsta-1 ,4-diene-17β-carbothioic acid S-fluoromethyl ester), or a pharmaceutically acceptable salt thereof.
The compound of the present invention will normally, but not necessarily, be formulated into pharmaceutical compositions prior to administration to a patient. Accordingly, in another aspect the invention is directed to pharmaceutical compositions comprising a compound of the invention and one or more pharmaceutically acceptable excipient.
The pharmaceutical compositions of the invention may be prepared and packaged in bulk form wherein a safe and effective amount of a compound of the invention can be extracted and then given to the patient, such as with powders or syrups. Alternatively, the pharmaceutical compositions of the invention may be prepared and
packaged in unit dosage form wherein each physically discrete unit contains a safe and effective amount of a compound of the invention. When prepared in unit dosage form, the pharmaceutical compositions of the invention typically contain from about 0.1 to 99.9 wt.%, depending on the nature of the formulation.
The pharmaceutical compositions of the invention typically contain one compound of the invention. However, in certain embodiments, the pharmaceutical compositions of the invention contain more than one compound of the invention. For example, in certain embodiments the pharmaceutical compositions of the invention contain two compounds of the invention. In addition, the pharmaceutical compositions of the invention may optionally further comprise one or more additional pharmaceutically active compounds.
As used herein, "pharmaceutically acceptable excipient" means a pharmaceutically acceptable material, composition or vehicle involved in giving form or consistency to the pharmaceutical composition. Each excipient must be compatible with the other ingredients of the pharmaceutical composition when commingled, such that interactions which would substantially reduce the efficacy of the compound of the invention when administered to a patient and would result in pharmaceutically unacceptable compositions are avoided. In addition, each excipient must of course be of sufficiently high purity to render it pharmaceutically acceptable.
The compound of the invention and the pharmaceutically acceptable excipient or excipients will typically be formulated into a dosage form adapted for administration to the patient by the desired route of administration. For example, dosage forms include those adapted for (1) inhalation, such as aerosols and solutions and (2) intranasal administration, such as solutions or sprays.
It will be appreciated by the skilled person that a preferred route of administration for treating asthma and COPD is inhalation and that a preferred route of administration for treating allergic rhinitis is intranasal administration.
Suitable pharmaceutically acceptable excipients will vary depending upon the particular dosage form chosen. In addition, suitable pharmaceutically acceptable excipients may be chosen for a particular function that they may serve in the composition. For example, certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the carrying or transporting the compound of the present invention once administered to the patient from one organ, or portion of the body, to another organ, or portion of the body. Certain pharmaceutically acceptable excipients may be chosen for their ability to enhance patient compliance.
Suitable pharmaceutically acceptable excipients include the following types of excipients: Diluents, fillers, binders, lubricants, glidants, granulating agents, solvents, co-solvents, suspending agents, emulsifiers, anticaking agents, humectants, chelating agents, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents. The skilled artisan will appreciate that certain pharmaceutically acceptable excipients may serve more than one function and may serve alternative functions depending on how much of the excipient is present in the formulation and what other ingredients are present in the formulation.
Skilled artisans possess the knowledge and skill in the art to enable them to select suitable pharmaceutically acceptable excipients in appropriate amounts for use in the invention. In addition, there are a number of resources that are available to the skilled artisan which describe pharmaceutically acceptable excipients and may be useful in selecting suitable pharmaceutically acceptable excipients. Examples include Remington's Pharmaceutical Sciences (Mack Publishing Company), Remington: The Science and Practice of Pharmacy, (Lippincott Williams & Wilkins), The Handbook of Pharmaceutical Additives (Gower Publishing Limited), and The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association and the Pharmaceutical Press).
The pharmaceutical compositions of the invention are prepared using techniques and methods known to those skilled in the art. Some of the methods commonly used in the art are described in Remington's Pharmaceutical Sciences (Mack Publishing Company).
Oral solid dosage forms such as tablets will typically comprise one or more pharmaceutically acceptable excipients, which may for example help impart satisfactory processing and compression characteristics, or provide additional desirable physical characteristics to the tablet. Such pharmaceutically acceptable excipients may be selected from diluents, binders, glidants, lubricants, disintegrants, colorants, flavorants, sweetening agents, polymers, waxes or other solubility- modulating materials.
Dosage forms for parenteral administration will generally comprise fluids, particularly intravenous fluids, i.e., sterile solutions of simple chemicals such as sugars, amino acids or electrolytes, which can be easily carried by the circulatory system and assimilated. Such fluids are typically prepared with water for injection USP. Fluids used commonly for intravenous (IV) use are disclosed in Remington, The Science and Practice of Pharmacy [full citation previously provided]. The pH of such IV fluids may vary, and will typically be from 3.5 to 8 as known in the art.
Dosage forms for nasal or inhaled administration may conveniently be formulated as aerosols, solutions, drops, gels or dry powders.
Dosage forms for nasal administration may be provided in a metered dose device. The dosage form may be provided as a fluid formulation for delivery from a fluid dispenser having a dispensing nozzle or dispensing orifice through which a metered dose of the fluid formulation is dispensed upon the application of a user-applied force to a pump mechanism of the fluid dispenser. Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid formulation, the doses being dispensable upon sequential pump actuations. The dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid formulation into the nasal cavity. In one embodiment, the fluid dispenser is of the general type described and illustrated in WO-A-2005/044354. The dispenser has a housing which houses a fluid discharge device having a compression pump mounted on a container for containing a fluid formulation. The housing has at least one finger-operable side lever which is movable inwardly with respect to the housing to cam the container upwardly in the housing to cause the pump to compress and pump a metered dose of the formulation out of a pump stem through a nasal nozzle of the housing. A particularly preferred fluid dispenser is of the general type illustrated in Figures 30-40 of WO-A-2005/044354.
For compositions suitable and/or adapted for inhaled administration, it is preferred that the compound or salt of formula (I) is in a particle-size-reduced form, and more preferably the size-reduced form is obtained or obtainable by micronisation. The preferable particle size of the size-reduced (e.g. micronised) compound or salt or solvate is defined by a D50 value of about 0.5 to about 10 microns (for example as measured using laser diffraction).
Aerosol formulations, e.g. for inhaled administration, can comprise a solution or fine suspension of the active substance in a pharmaceutically acceptable aqueous or non-aqueous solvent. Aerosol formulations can be presented in single or multidose quantities in sterile form in a sealed container, which can take the form of a cartridge or refill for use with an atomising device or inhaler. Alternatively the sealed container may be a unitary dispensing device such as a single dose nasal inhaler or an aerosol dispenser fitted with a metering valve (metered dose inhaler) which is intended for disposal once the contents of the container have been exhausted.
Where the dosage form comprises an aerosol dispenser, it preferably contains a suitable propellant under pressure such as compressed air, carbon dioxide or an organic propellant such as a hydrofluorocarbon (HFC). Suitable HFC propellants include 1 , 1 ,1 , 2,3,3, 3-heptafluoropropane and 1 ,1 ,1 ,2-tetrafluoroethane. The aerosol dosage forms can also take the form of a pump-atomiser. The pressurised aerosol may contain a solution or a suspension of the active compound. This may require the incorporation of additional excipients e.g. co-solvents and/or surfactants to improve
the dispersion characteristics and homogeneity of suspension formulations. Solution formulations may also require the addition of co-solvents such as ethanol. Other excipient modifiers may also be incorporated to improve, for example, the stability and/or taste and/or fine particle mass characteristics (amount and/or profile) of the formulation.
For pharmaceutical compositions suitable and/or adapted for inhaled administration, it is preferred that the pharmaceutical composition is a dry powder inhalable composition. Such a composition can comprise a powder base such as lactose, glucose, trehalose, mannitol or starch, the compound of formula (I) or salt or solvate thereof (preferably in particle-size-reduced form, e.g. in micronised form), and optionally a performance modifier such as L-leucine or another amino acid, and/or metals salts of stearic acid such as magnesium or calcium stearate. Preferably, the dry powder inhalable composition comprises a dry powder blend of lactose and the compound of formula (I) or salt thereof. The lactose is preferably lactose hydrate e.g. lactose monohydrate and/or is preferably inhalation-grade and/or fine-grade lactose. Preferably, the particle size of the lactose is defined by 90% or more (by weight or by volume) of the lactose particles being less than 1000 microns (micrometres) (e.g. 10-1000 microns e.g. 30-1000 microns) in diameter, and/or 50% or more of the lactose particles being less than 500 microns (e.g. 10-500 microns) in diameter. More preferably, the particle size of the lactose is defined by 90% or more of the lactose particles being less than 300 microns (e.g. 10-300 microns e.g. 50-300 microns) in diameter, and/or 50% or more of the lactose particles being less than 100 microns in diameter. Optionally, the particle size of the lactose is defined by 90% or more of the lactose particles being less than 100-200 microns in diameter, and/or 50% or more of the lactose particles being less than 40-70 microns in diameter. Most importantly, it is preferable that about 3 to about 30% (e.g. about 10%) (by weight or by volume) of the particles are less than 50 microns or less than 20 microns in diameter. For example, without limitation, a suitable inhalation-grade lactose is E9334 lactose (10% fines) (Borculo Domo Ingredients, Hanzeplein 25, 8017 JD Zwolle, Netherlands).
Optionally, in particular for dry powder inhalable compositions, a pharmaceutical composition for inhaled administration can be incorporated into a plurality of sealed dose containers (e.g. containing the dry powder composition) mounted longitudinally in a strip or ribbon inside a suitable inhalation device. The container is rupturable or peel-openable on demand and the dose of e.g. the dry powder composition can be administered by inhalation via the device such as the DISKUS ™ device, marketed by GlaxoSmithKline. The DISKUS ™ inhalation device is for example described in GB 2242134 A, and in such a device at least one container for the pharmaceutical composition in powder form (the container or containers preferably being a plurality of sealed dose containers mounted longitudinally in a strip or ribbon) is defined between two members peelably secured to one another; the device comprises: a means of defining an opening station for the said container or containers; a means
for peeling the members apart at the opening station to open the container; and an outlet, communicating with the opened container, through which a user can inhale the pharmaceutical composition in powder form from the opened container.
It will be appreciated that when the compound of the present invention is administered in combination with other therapeutic agents normally administered by the inhaled, intravenous, oral or intranasal route, that the resultant pharmaceutical composition may be administered by the same routes.
The compound of the present invention may conveniently be administered in amounts of, for example, 1 μg to 100 mg. The precise dose will of course depend on the age and condition of the patient and the particular route of administration chosen.
Biological test methods
Compounds of the invention may be tested for in vitro activity in accordance with the following assays:
1. Receptor Assay - Time-resolved fluorescence resonance energy transfer kinase assay
Recombinant human Syk was expressed as a His-tagged protein*. The activity of Syk was assessed using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay.
Version A - 3 μl of substrate reagent containing biotinylated peptide, Biotin- AAAEEIYGEI (0.5μM final), ATP (30μM final) and MgCI2 (1OmM final) in HEPES pH 7.4, (40 mM final), were added to wells containing 0.2μl of various concentrations of compound or DMSO vehicle (3.3% final) in Greiner low volume 384 well black plate. The reaction was initiated by the addition of 3μl of Syk (2OnM final) in HEPES pH 7.4 (40μM final). The reaction was incubated for 40 minutes at room temperature, then terminated by the addition of 3μl of read reagent containing 60 mM EDTA, 15OmM NaCI, 5OnM Streptavidin APC (Prozyme, San Leandro, California, USA), 0.5nM antiphosphotyrosine antibody labelled with W-1024 europium chelate (Wallac OY, Turku, Finland) in 4OmM HEPES pH 7.4. The reaction was further incubated for 60 minutes at room temperature. The degree of phosphorylation of Biotin-AAAEEIYGEI was measured using a BMG Rubystar plate reader (BMG LabTechnologies Ltd, Aylesbury, UK) as a ratio of specific 665 nm energy transfer signal to reference europium 620 nm signal.
Version B - 3 μl of substrate reagent containing biotinylated peptide, Biotin- AAAEEIYGEI (0.5μM final), ATP (30μM final) and MgCI2 (13mM final) in HEPES pH 7.4, (40 mM final), were added to wells containing 0.1 μl of various concentrations of compound or DMSO vehicle (1.7% final) in Greiner low volume 384 well black plate. The reaction was initiated by the addition of 3μl of Syk (2nM final) in HEPES pH 7.4 (4OmM final). The reaction was incubated for 60min at room temperature, then terminated by the addition of 3μl of read reagent containing 60 mM EDTA, 15OmM NaCI, 5OnM Streptavidin APC (Prozyme, San Leandro, California, USA), 0.5nM antiphosphotyrosine antibody labelled with W-1024 europium chelate (Wallac OY, Turku, Finland) in 4OmM HEPES pH 7.4. The reaction was further incubated for 45min at room temperature. The degree of phosphorylation of Biotin-AAAEEIYGEI was measured using a BMG Rubystar plate reader (BMG LabTechnologies Ltd, Aylesbury, UK) as a ratio of specific 665 nm energy transfer signal to reference europium 620 nm signal.
Compounds according to the present invention were assayed in this, or a similar Time-resolved fluorescence resonance energy transfer kinase assay, and gave IC50 values less than 10μM.
* Preparation of Recombinant Human Full Length Spleen Tyrosine Kinase (Svk)
Full length human Syk was expressed with a 6His tag on the N-terminal using the baculovirus system (Invitrogen, Paisley, Scotland). The cells were disrupted by dounce homogenisation, the debris removed by centrifugation and the lysate contacted with NiNTA Superflow (Qiagen, Crawley, UK). The NiNTA was packed into a column and eluted using 10 column volumes each of buffer (2OmM Tris pH8.0, 30OmM NaCI, 1OmM βMcEtOH, 10% glycerol), buffer + 1 M NaCI, buffer + 2OmM Imidazole and buffer + 30OmM imidazole. The 30OmM Imidazole fractions were pooled buffer exchanged using G25M (Amersham Biosciences, Buckinghamshire, UK) into 2OmM MES pH 6.0, 2OmM NaCI, 1OmM βMcEtOH,10% glycerol. The buffer exchanged 6His-Syk was loaded onto a Source15S column (Amersham Biosciences, Buckinghamshire, UK) and the column eluted using a NaCI gradient 0-50OmM over 50 column volumes. The 6His-Syk containing fractions were pooled and concentrated by ultra-filtration. The identity of 6His-Syk was confirmed by peptide mass finger printing and intact LC-MS.
2. Whole Cell Assay - cFms assay
Principle of the assay
Cells of the mouse fibroblast cell line NIH-3T3 are stably transfected with a cFms- SYK chimera. Addition of the ligand (MCSF) produces dimerisation of the chimera resulting in autophosphorylation of the SYK kinase domain. Following cell lysis phosphorylated SYK is detected by ELISA.
Stimulation of cFms-SYK cells with MCSF
Cells are plated at a density of 1x105/well in a volume of 200μL growth medium (DMEM containing 10% heat inactivated foetal calf serum, 1% L-glutamine, 400μg/mL geneticin and 400μg/mL zeocin) in 96 well Collagen 1 coated tissue culture plates. Following incubation at 370C, 10% CO2, for 20 hours the cell supernatant is removed and replaced with 200μL DMEM containing 1 % penicillin/streptomycin (serum free DMEM). The cells are incubated for one hour under the conditions described above. The medium is removed, 50μL appropriately diluted compound solution added and the plate incubated for a further hour. Cells are stimulated with 25μL MCSF (0.66μg/mL final) for 20 minutes at 37°C. After removal of the supernatant, the cells are washed with cold PBS and lysed with 100μL lysis buffer for 4 hours at 40C.
cFms ELISA
85μL cell lysate is transferred to a 96 well ELISA plate coated with goat anti human M-CSF R capture antibody and incubated for 16 hours at 4°C. The plate is washed and a biotinylated anti-phosphotyrosine detection antibody added (100μLJwell) for 2 hours at room temperature. This is removed and replaced with 100μl_ Streptavidin- HRP for 30minutes. Captured phosphorylated SYK is visualised using 100μL TMB substrate. The reaction is terminated with 50μL 1M sulphuric acid and the absorbance measured at 450nm.
Compound Preparation
Compound is prepared as a 1OmM stock in DMSO and a dilution series prepared in DMSO using 9 successive 5-fold dilutions. This dilution series is diluted a further 1 :333 with serum free DMEM to give the concentration range to be tested of 1x10"5 to 1.54x10"11M. Compound dilutions are prepared using the Biomek 2000 or Biomek Nx automated robotic pipetting systems.
3. B Cell Proliferation Assay
Background
The population of B cells observed in this assay are the naive mature IgM/lgD expressing population. These form at least 70% of the purified B cell population (the rest being isotype switched memory B cells) and are the only cells that proliferate as the cells are stimulated with anti-lgM.
Anti-lgM drives signalling through the B cell receptor which is Syk dependant. Proliferation is a functional measure of B cell signalling that can be measured by observing the incorporation of tritiated methyl thymidine into the cells.
Protocol
Purified human tonsillar B cells are resuspended in Buckleys* medium at a concentration of 1.25 x 106 ml.
160μl of cells re-suspended in Buckley's medium is added to the compound and control wells of a 96 well plate. The control wells are located on column 11 and 12 of the 96 well plate. The background wells are located in column 12 and 20μl of 10μM control is added to provide an appropriate background control. 20μl of 1% DMSO is added to the wells in column 11 for the stimulated control.
The compound titrations are located between columns 1 and 10. Three compounds are run in duplicate on each plate and row A and B are used for the control compound titration.
The final concentration of DMSO is 0.1% in the assay. The cells are left for 45 minutes, after 45 minutes the proliferative stimulus is added to the first 11 wells of the
96 well plate and 20μl of medium is added to column 12. F(ab')2 fragments of a polyclonal goat anti-sera raised to human IgM is used at a final concentration of 15μg/ ml to stimulate the cells. (Biosource. Cat no: AMI 4601 ).
Tritiated methyl thymidine is added to the cells at a concentration of 1 μCi per well. (Amersham, TRK 758). The radioactivity is added 65 hours after the initial stimulus and is left on the cells for 6 to 8 hours. After pulsing with methyl thymidine the cells are harvested on a Skatron 96 well cell harvester onto glass fibre mats. Once these have dried these are counted on a Wallac 1450 Microbeta scintillation counter.
Data is downloaded as an XL file and IC50's determined using Activity base.
* Buckleys Medium: 450 mis Iscoves (Sigma I 3390), 5OmIs FCS, 2.5 g BSA, 5mls Pen/ strep, 5mls Glutamine (20OmM), 500μl Apo transferrin (50mg/ml) Sigma (T 1147), 100μl Bovine Insulin (10mg/ml) Sigma (1 1882).
Compound Preparation
Compound is prepared as a 1OmM stock in DMSO and a dilution series prepared in DMSO using 9 successive 3-fold dilutions. This dilution series is diluted a further 1 :100 with Buckleys medium to give the concentration range to be tested of 100μM to 5nM. This is added as 20μl to 96 well plates in duplicate to generate two IC50's for each compound tested. Each plate is run in the presence of a control compound, which acts as an internal standard. .
4. LAD2 Assay
Principle of the assay
LAD2 is a stem cell factor (SCF)-dependent human mast cell line that was established by the NIH from bone marrow aspirates from a patient with mast cell sarcoma/leukaemia. LAD2 cells resemble CD34+-derived human mast cells and express functional FcεRl. The FcεRI is up-regulated in the presence of IL-4, SCF and IgE, subsequent cross linking of cell-bound IgE results in degranulation which can be measured as hexosaminidase release.
Priming LAD2 cells to up-regulate FcεRI
LAD2 cells are re-suspended at 1x105/ml in complete stem pro-34SFM (Gibco Cat 10640-019 media containing Stem Pro-34 nutrient supplement (1 :40), glutamine (2mM), penicillin (100u/ml), streptomycin (100μg/ml)) with additional supplements of human recombinant SCF (100ng/ml; R&D systems), human recombinant Interleukin- 4 (6ng/ml; R&D Systems) and IgE (100μg/ml; Calbiochem). Cells are then maintained for 5 days at 370C, 5% CO2 in a humidified atmosphere.
Compound Preparation
Compounds are titrated from a 2mM stock in 100% DMSO to give 9 successive 1 :3 dilutions (V 96-well Nunc; Biomek 2000). From this master plate 3μl is dispensed into a daughter plate (flat 96-well NuncBiomek Fx) which is then diluted 1:40 in RPMI with 2mM glutamine, and 20μl of the diluted compound transferred into the Greiner cell plate. Therefore the final compound concentration range is IxIO-5M to 5x10"10M in a constant 0.5% DMSO. Control wells are treated with 0.5% DMSO.
Activation of LAD2 cells with anti-lgE
Primed LAD2 cells are centrifuged (30Og, 5min), the supernatant discarded and the cell pellet re-suspended at 1x104 cells/ml in RPMI supplemented with glutamine (2mM). Following a further centrifugation (30Og, 5min) the cells are re-suspended in fresh RPMI with glutamine (2mM), adjusted to a density of 2.85x105/ml, and pipetted into sterile V-well plates (70μl_/well; Greiner) containing 20μl diluted compound (prepared as detailed above). Cells are then incubated for 1h (370C, 5% CO2 in a humidified atmosphere) before activating with a sub-maximal concentration of anti- lgE (10μl volume to give a final assay dilution of 1 :2700; Sigma). Following a 40 min incubation (370C, 5% CO2 in a humidified atmosphere), plates are centrifuged (120Og, 10 min, 40C) and the supernatant removed for hexosaminidase assay. The cell pellet is lysed in 100μl/well triton-X (0.5% in RPMI 2mM glutamine) at 370C for 30 minutes.
Beta-hexosaminidase assay
Beta-hexosaminidase activity is measured by the conversion of 4-methylumbelliferyl N-acetyl-ε-D glucosaminide (Sigma) to a fluorescent product.
Supernatant or lysate (25μL) is incubated with an equal volume of 4- methylumbelliferyl N-acetyl-ε-D glucosaminide (500μM in 0.2M sodium citrate buffer, pH 4.5) in black 96-well plate (Nunc) for 1 hour at 370C. The reaction is then terminated by addition of Trizma pH9 (90μL) and the fluorescent product measured using excitation 356nm and emission 450nm (Tecan Safire)
A useful screening strategy comprises assay 1 (enzyme assay (pKi), assay 2 and then assay 3 (B Cell Proliferation) or assay 4 (LAD2).
Intermediates and Examples
General
All temperatures are in 0C.
DMSO refers to dimethylsulfoxide.
DMF refers to Λ/,Λ/-dimethylformamide
THF refers to tetrahydrofuran.
HPLC refers to high performance liquid chromatography.
ADDP refers to 1 ,1'(azodicarbonyl)dipiperidine
SPE refers to solid phase extraction cartridges marketed by lsolute
TBTU refers to O-benzotriazol-1-yl-Λ/,Λ/,Λ/',Λ/',-bis(tetramethylene)uronium tetrafluoroborate
HOBt refers to N-hydroxybenzotriazole hydrate
1H NMR spectra were recorded using a Bruker DPX 400MHz, a Bruker AMX 300 or a Bruker AMX 500 spectrometer referenced to tetramethylsilane. Fluorine NMR (282 MHz) spectra were obtained on a Bruker AMX 300 spectrometer.
"Hydrophobic frits" refers to filtration tubes sold by Whatman. SPE (solid phase extraction) refers to the use of cartridges sold by International Sorbent Technology Ltd.
TLC (thin layer chromatography) refers to the use of TLC plates sold by Merck coated with silica gel 60 F254.
For Examples 1 to 84
LCMS was conducted on a Supelcosil LCABZ+PLUS column (3.3 cm x 4.6 mm ID) eluting with 0.1% HCO2H and 0.01 M ammonium acetate in water (solvent A) and 0.05% HCO2H 5% water in acetonitrile (solvent B), using the following elution gradient 0.0-7min 0%B, 0.7-4.2 min 100%B, 4.2-5.3 min 0%B, 5.3-5.5min 0%B at a flow rate of 3ml/min. The mass spectra were recorded on a Fisons VG Platform spectrometer using electrospray positive and negative mode (ES+ve and ES-ve).
Flashmaster refers to an automated multi-user flash chromatography system, available from Argonaut Technologies Ltd, which utilises disposable, normal phase, SPE cartridges (2 g to 100 g). It provides quaternary on-line solvent mixing to enable gradient methods to be run. Samples are queued using the multi-functional open access software, which manages solvents, flow-rates, gradient profile and collection conditions. The system is equipped with a Knauer variable wavelength uv-detector and two Gilson FC204 fraction-collectors enabling automated peak cutting, collection and tracking.
Preparative mass directed HPLC was conducted on a Waters FractionLynx system comprising of a Waters 600 pump with extended pump heads, Waters 2700 autosampler, Waters 996 diode array and Gilson 202 fraction collector on a 10 cm 2.54 cm ID ABZ+ column, eluting with either 0.1% formic acid or trifluoroacetic acid in water (solvent A) and 0.1% formic or trifluoroacetic acid in acetonitrile (solvent B) using the appropriate elution gradient. Mass spectra were recored on Micromass ZMD mass spectrometer using electrospray positive and negative mode, alternate scans. The software used was MassLynx 3.5 with OpenLynx and FractionLynx options.
Intermediates
Intermediate 1 : 2,3-dihvdro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide

Method 1
To a mixture of 6-amino-1 ,2-benzisothiazol-3(2H)-one 1 ,1 -dioxide (6.37g) in cone hydrochloric acid (79.6ml) was added zinc dust (18.9g) partitioned over half an hour cooling with an ice bath when necessary (maximum temperature 7O0C). The reaction was stirred at room temperature for 3h. The mixture was basified with a solution of saturated NaHCO3 (50ml), then with solid NaHCO3. The mixture was filtered and the filtrate was extracted with ethyl acetate (3x150ml). The organic phases were combined, dried (brine and MgSO4), filtered and concentrated under vacuum to give a pale yellow solid (1.548g). The residue from filtration of the basified reaction was triturated with ethyl acetate (500ml) and filtered. The orange filtrate was concentrated under vacuum to give an orange solid (1.789g). The residue obtained from filtration was triturated again with ethyl acetate (250ml), filtered and concentrated under vacuum to give a pale orange solid (0.357g). All the solids were dissolved in methanol, combined and then concentrated to give the desired product as a beige solid (3.151g). LC/MS: Rt 0.83min.
Method 2

400 MHz NMR in dimethyl sulphoxide-d6. Dimethyl sulphoxide-d5 as reference at 2.50 ppm. δ (ppm): 1.30 (3) t J=7.1Hz, 4.32 (2H) q J=7.1Hz, 5.01 (2H) s, 7.64-7.70 (2H) m, 7.81 (1 H) td J=7.6Hz J=1.0Hz, 7.98 (1 H) d J=8.0Hz

This was suspended in acetonitrile (400ml) and water (320ml) at almost reflux, treated with further acetonitrile (40ml), and aged for 1h. The mixture was cooled slowly to ambient temperature over 3h, cooled further to 0-50C and aged for 30min. The product was isolated by filtration, washed with 1 :1 wateπacetonitrile (140ml), then water (140ml), and dried in vacuo to give the title compound as an off-whte solid (64.5g).
400 MHz NMR in dimethyl sulphoxide-d6. TMS as reference at 0.00 ppm. δ (ppm): 1.31 (3H) t J=7.1 Hz, 4.35 (2H) q J=7.1Hz, 5.16 (2H) s, 7.96 (1 H) dq J=8.6Hz
J=0.5Hz, 8.61 (1 H) dd J=8.6Hz J=2.2Hz, 8.90 (1H) d J=2.1 Hz
Step 3
A suspension of the above 6-nitro-2,3-dihydro-1 ,2-benzisothiazol-1,1 -dioxide (10.0g), 5%Pd/C (1.0g, 50%wet) and methanesulfonic acid (4.5ml) in propan-2-ol (100ml) was hydrogenated under hydrogen at 2bar pressure at 5O0C. The mixture was purged with nitrogen, cooled to 2O0C and treated with a mixture of 2M sodium hydroxide (90ml) and 10M sodium hydroxide (10ml). The reaction was stirred for 2h
at ambient temperature and filtered through a filter pad, washing the pad with water (20ml). 5M hydrochloric acid (45ml) was added to the filtrate to adjust to pH7. The resultant slurry was stirred at ambient temperature for 2h, cooled to 0-50C and aged for a further 1h. The product was isolated by filtration, washed with cold 2:1 water:propan-2-ol (30ml) then water (2x30ml). The solid was dried in vacuo to give the title compound as a light grey solid (5.23g).
400 MHz NMR in dimethyl sulphoxide-d6. Dimethyl sulphoxide-d5 as reference at 2.50 ppm. δ (ppm): 4.18 (2H) s, 5.59 (2H) s, 6.79 (1 H) d J=2.1 Hz, 6.83 (1 H) dd J=8.3Hz J=2.1Hz, 7.14 (1H) d J=8.3Hz, 7.52 (1 H) br s
Intermediate 2: 5-amino-1 ,2-benzisothiazol-3(2H)-one 1 ,1 -dioxide

A vigorously stirred solution of 5-nitro-1 ,2-benzisothiazol-3(2H)-one 1 ,1 -dioxide (for preparation see Journal of Heterocyclic Chemistry 1986, 23(4), 1253-5) (1.99g) in ethanol (80ml) was hydrogenated at room temperature and 1 atmosphere of pressure using palladium on carbon catalyst (400mg) for 2 days. The mixture was filtered and the solvent was evaporated in vacuo before the residue was re-dissolved in ethanol (80ml) and was hydrogenated at room temperature and 1 atmosphere of pressure using palladium on carbon catalyst (400mg) for a further one day. The mixture was filtered and the solvent was evaporated in vacuo before the residue was dissolved in a small quantity of methanol, and silica gel was added to form a slurry. The slurry had the solvent evaporated in vacuo and the solid was placed onto the top of a 5Og silica cartridge. A frit was placed onto the top of the solid before the cartridge was eluted using a 0-15% gradient of (1% triethylamine in methanol) in dichloromethane. The required fractions were combined and the solvent was evaporated in vacuo to give the title compound as a brown oil (603mg). LC/MS: Rt 1.68 min, [M-HH 97.
Intermediate 3: 2,3-dihvdro-1,2-benzisothiazol-5-amine 1 ,1-dioxide:

Zinc dust (2.4g) was added portion wise to a stirred suspension of 5-amino-1 ,2- benzisothiazol-3(2H)-one 1 ,1-dioxide (820mg) in concentrated hydrochloric acid
(10ml). The mixture was stirred at room temperature for 20 hours before saturated aqueous sodium hydrogen carbonate solution was added to the mixture until the pH of the solution was 8. The mixture was filtered and extracted with ethyl acetate (4x150ml). The combined organic phases were dried (MgSO4), filtered and the solvents evaporated in vacuo to give the title compound as a yellow solid (230mg). LC/MS: Rt 0.82min, MH+185.
Intermediate 4: 2-bromo-2,2-difluoro-Λ/-(2-hydroxy-5-nitrophenyl)acetamide
Triethylamine (1.97g, 19.46mmol) was added to an ice-cooled solution of 2-amino-4- nitrophenol (3g, 19.46mmol) in THF (30ml). Bromodifluoroacetylbromide (2.48ml, 19.46mmol) was added dropwise to the solution and the mixture stirred at O0C for 45min. The reaction was quenched with water and extracted with ethyl acetate (3x 50ml). The combined organic extracts were washed with brine, dried (magnesium sulphate) and reduced to dryness under vacuum to give 2-bromo-2,2-difluoro-/V-(2- hydroxy-5-nitrophenyl)acetamide (7.09g) as a brown solid. LC/MS: Rt 3.09min, [M- H]- 311/309.
Intermediate 5: 2,2-difluoro-6-nitro-2H-1 ,4-benzoxazin-3(4H)-one
2-bromo-2,2-difluoro-Λ/-(2-hydroxy-5-nitrophenyl)acetamide (5.6g, 18mmol) and potassium carbonate (7.46g, 54mmol) in DMF (50ml) were stirred for 18hrs at 5O0C. The reaction was filtered and the filtrate reduced to dryness under vacuum. The residue was partitioned between ethyl acetate and water and the aqueous extracted with ethyl acetate (x3). The combined organic phases were washed with a little water and the solvent evaporated under vacuum to give 2,2-difluoro-6-nitro-2H-1 ,4- benzoxazin-3(4/-/)-one as a sticky yellow solid, which was used without further purification. LC/MS: Rt 2.86min, [M-H]" 229.
Intermediate 6: 6-amino-2,2-difluoro-2H-1 ,4-benzoxazin-3(4/-/)-one
2,2-difluoro-6-nitro-2H-1,4-benzoxazin-3(4H)-one (2.87g, 12.47mmol) and palladium on carbon (10%, 650mg) in ethyl acetate (100ml) were hydrogenated under 1Atm. of hydrogen for 5hrs. The reaction was filtered through celite, the residue washed with ethyl acetate and the filtrate and washings reduced to dryness under vacuum. The crude product was purified by chromatography on a Flashmaster cartridge (Si, 7Og), eluting with a MeOH/DCM/triethylamine gradient (0-15% MeOH, 1% triethylamine) and then washing the column with further MeOH/DCM/triethylamine (10% MeOH, 1% triethylamine. The product fractions were reduced to dryness under vacuum to give 6-amino-2,2-difluoro-2H-1 ,4-benzoxazin-3(4H)-one (857mg) as a brown solid. LC/MS: Rt 2.25min, [M-H]- 199.
Intermediate 7: ethyl r(3-nitrophenvl)oχylacetate
A mixture of 3-nitrophenol (6.37g, 45.8mmol), ethyl bromoacetate (6.09ml, 54.96ml) and potassium carbonate (12.7g, 91.6mmol) in DMF (40ml) were stirred for 3hrs at 7O0C. The reaction was filtered and the filtrate reduced to dryness under vacuum. The residue was partitioned between ethyl acetate and water and the aqueous extracted with ethyl acetate. The combined organic phases were washed with a little water and the solvent evaporated under vacuum. The residue was purified by chromatography on Flashmaster a cartridge (Si, 100g) eluting with an ethyl acetate / cyclohexane gradient (0-50%). The product fractions were reduced to dryness under vacuum to give ethyl [(3-nitrophenyl)oxy]acetate (9.63g) as a yellow oil. LC/MS: Rt 2.95min, MH+ not observed.
Intermediate 8: Λ/-methyl-2-f(3-nitrophenyl)oxylacetamide
Ethyl [(3-nitrophenyl)oxy]acetate (4g, 17.76mmol) in methylamine in methanol (2M, 54ml) was stirred at room temperature overnight. The reaction was filtered, the solid washed with methanol and dried to give Λ/-methyl-2-[(3-nitrophenyl)oxy]acetamide (1.76g) as a white solid. Evaporation of the filtrate and trituration of the residue with a minimal quantity of methanol gave a further 1.31g of the desired product. LC/MS: Rt 2.28min, MH+ 211.
Intermediate 9: 2-r(3-aminophenyl)oxyl-Λ/-methylacetamide
Λ/-methyl-2-[(3-nitrophenyl)oxy]acetamide (3.07g, 14.6mmol), and palladium on carbon (10%, 700mg) in ethanol (100ml) were hydrogenated under 1Atm. of hydrogen for 5hrs. The reaction was filtered through celite, the residue washed with ethyl acetate and the filtrate and washings reduced to dryness under vacuum to give 2-[(3-aminophenyl)oxy]-Λ/-methylacetamide as a grey/white solid (2.62g). LC/MS: Rt 0.86min, MH+ 181.
Intermediate 10: r3-(4-methyl-1 ,3-oxazol-5-yl)phenyllamine
A solution of 4-methyl-5-(3-nitrophenyl)-1 ,3-oxazole (5Og) in methanol (600ml) was prepared in parr-shaker bottle; Pd/C (5g) was transferred carefully and hydrogenated at 3.5kg pressure for 20hr. The reaction mixture was filtered over celite bed and the filtrate was concentrated. The residue was purified by silica gel column chromatograph using 40% ethyl acetate in pet.ether to get [3-(4-methyl-1 ,3-oxazol-5- yl)phenyl]amine as a yellow coloured solid. Yield 18g (42%).
Intermediate 11: 4-methyl-5-(3-nitrophenyl)-1,3-oxazole
To a cooled (0 0C) solution of 1-isocyanoethyl 4-methylphenyl sulfone (6Og, 0.287mol) in dry methanol (800ml), dry powdered potassium carbonate was added and stirred at RT for 1 hr. The reaction was recooled to 0 0C and 3-nitrobenzaldehyde added. The reaction mixture was warm to RT, followed by heating at 55 0C overnight
with stirring. The excess solvent was removed under reduced pressure and the residue was dissolved in water (100ml). The mixture was extracted with ethyl acetate (3 x 350ml). The combined organic layer was washed with brine, water, dried over anhydrous sodium sulfate and concentrated. The crude product was purified by silica get (60-120mesh) column chromatograph using 15% ethyl acetate in pet.ether to give 4-methyl-5-(3-nitrophenyl)-1 ,3-oxazole as yellow coloured solid. Yield 5Og (86%).
Intermediate 12: 1-isocvanoethyl 4-methylphenyl sulfone
To cooled (0 0C) solution of {1-[(4-methylphenyl)sulfonyl]ethyl}formamide (110g, 0.484mol) in dry THF (1000ml), POCI3 (148.4g, 0.968mol) was added slowly and stirred for half an hour. Then triethyl amine (293.8g, 2.9mol) was added in drops for half an hour and stirred the reaction mixture for 2hr at 0 0C. TLC was checked, there is no starting material and reaction mixture was quenched with water. The mixture was extracted with ethyl acetate (3 x 350ml). The combined organic layer was washed with brine, water, dried over anhydrous sodium sulfate and concentrated. The crude 1-isocyanoethyl 4-methylphenyl sulfone was taken to next step without further purification. Yield (crude): 6Og.
Intermediate 13: {1-r(4-methylphenyl)sulfonyl1ethyl)formamide
A stirred mixture of dry acetonitrile (500ml) and dry toluene (500ml) was cooled to - 30 0C, followed by the addition of formamide (50.7g, 1.1 mol), acetaldehyde (37g, 0.85mol) and TMS-CI (106.4g, 0.98mol) at -30 0C under nitrogen atmosphere. The reaction mixture was stirred at RT for 1 hr and warm to 55 °C, then para-toluene sulphonic acid was added and stirred the reaction mixture at this temperature overnight. The reaction mixture was cooled to 0 0C, tert-butyl ether (300ml) added and the mixture stirred for 1hr. The white precipitate formed was filtered off and dried. The crude {1-[(4-methylphenyl)sulfonyl]ethyl}formamide was taken into next step without further purification. Yield (crude): 11Og
Intermediate 14: A/-(2-chloro-4-pyrimidinyl)-1/-/-indazol-4-amine
A mixture of 1 H-indazol-4-ylamine (2.72 g) (Intermediate 15), sodium bicarbonate (6.87 g) and 2,4-dichloropyrimidine (9.14 g) in 2-propanol (110 mL) was stirred at reflux overnight. The reaction mixture was cooled and the precipitate filtered, washed with ether and then the solid extracted with methanol. The methanolic solution was evaporated in vacuo to give the title compound as a pale-brown solid. LC/MS: Rt 2.6min.
Intermediate 15: 5-methyl-4-nitroindazole
To a solution of 2,4-dimethyl-3-nitroaniline (6.0 g) (Synthesis 10, 761-3 (1989)) in acetic acid (180 ml) was added f-butyl nitrite dropwise at 15 0C. This was left to stir at room temperature for 1.5 hours. The reaction mixture was evaporated and partitioned between ethyl acetate (150 ml) and saturated sodium bicarbonate solution (100 ml). The organic layers were combined, dried and evaporated to leave a red solid. LC/MS: Rt 2.84 min.
Intermediate 16: Λ/-[2-(diethylamino)ethyll-3-nitrobenzenesulfonamide
N,N,N'-diethylethylenediamine (Aldrich, 2.1g, 18.0mmol) was added to an ice-cooled solution of 3-nitrobenzenesulphonylchloride (2g, 9.0mmol) in DCM (15ml) and the reaction stirred at room temperature for 17hrs. Water (15ml) was added to the mixture, the phase separated (frit) and the organic phase applied directly to a biotage column (Si, 4Og). The column was eluted with DCM, ethanol, ammonia (200:8:1), to give, after evaporation of the solvents, Λ/-[2-(diethylamino)ethyl]-3- nitrobenzenesulfonamide (1.82g) as a yellow oil. LC/MS: MH+ 302.
Intermediate 17: 3-amino-N-r2-(diethylamino)ethyllbenzenesulfonamide
Iron powder (1.22g, 22.0mmol) was added in portions to an ice-cooled solution of N- [2-(diethylamino)ethyl]-3-nitrobenzenesulfonamide (1.66g, 5.5mmol) in acetic acid (60ml). The reaction was stirred at room temperature for 18hrs, diluted with water (250ml) and ethyl acetate (100ml) and solid sodium hydrogen carbonate added to the mixture until pH7 was achieved. The organic phase was separated, the aqueous extracted with ethyl acetate (3x 100ml). The combined organic phases were washed with water and brine, dried (magnesium sulphate) and reduced to dryness in vacuo. The residue was purified by chromatography on a biotage system (Si) eluting with DCM, ethanol, ammonia (200:8:1 - 100:8:1), to give, after evaporation of the solvents, 3-amino-N-[2-(diethylamino)ethyl]benzenesulfonamide as a pale yellow oil (0.44g, 29%). LC/MS: MH+ 272.
Intermediate 18: Λ/-r2-(4-morpholinyl)ethyll-3-nitrobenzenesulfonamide
4-(2-aminoethyl)morpholine (Aldrich, 2.3g, 18.0mmol) was added to an ice-cooled solution of 3-nitrobenzenesulphonylchloride (2g, 9.0mmol) in DCM (15ml) and the reaction stirred at room temperature for 17hrs. Water (15ml) was added to the mixture, the phase separated (frit) and the organic phase applied directly to a biotage column (Si, 40g). The column was eluted with DCM, ethanol, ammonia (200:8:1), to give, after evaporation of the solvents Λ/-[2-(4-morpholinyl)ethyl]-3- nitrobenzenesulfonamide (1.92g, 67%) as an off-white solid. LC/MS: MH+ 316.
Intermediate 19: 3-amino-N-r2-(4-morpholinyl)ethvπbenzenesulfonarnide
Iron powder (1.34g, 24.1mmol) was added in portions to an ice-cooled solution of N- [2-(4-morpholinyl)ethyl]-3-nitrobenzenesulfonamide (1.9Og, 6.0mmol) in acetic acid (60ml). The reaction was stirred at room temperature for 18hs, diluted with water (250ml) and ethyl acetate (100ml) and solid sodium hydrogen carbonate added to the mixture until pH7 was achieved. The organic phase was separated, the aqueous extracted with ethyl acetate (3x 100ml). The combined organic phases were washed with water and brine, dried (magnesium sulphate) and reduced to dryness in vacuo. The residue was purified by chromatography on a biotage system (Si) eluting with DCM, ethanol, ammonia (200:8:1), to give, after evaporation of the solvents, 3- amino-N-[2-(4-morpholinyl)ethyl]benzenesulfonamide as a pale yellow oil (0.8g, 47%). LC/MS: MH+ 286.
Intermediate 20: 1 -(3-nitrophenyl)-/V-(3-f(triethylsilyl)oxylpropyl)methanesulfonamide
To an ice-cold solution of 3-nitrophenylmethanesulfonyl chloride (Alfa Aesar, 0.5Og, 2.12mmol) in methylene chloride (8ml) was added a solution of {3- [(triethylsilyl)oxy]propyl}amine (0.44g, 2.33mmol) in methylene chloride (2ml), followed by the dropwise addition of Λ/,Λ/-diisopropylethylamine (0.3Og, 2.33mmol). The reaction mixture was stirred with gradual warming to room temperature, under nitrogen, for 2Oh. The reaction mixture was diluted with methylene chloride (100ml), and washed with water (2 x 50ml) and brine (50ml). The organic layer was dried (sodium sulfate) and concentrated under reduced pressure to afford 1-(3- nitrophenyl)-Λ/-{3-[(triethylsilyl)oxy]propyl}methanesulfonamide (0.67g, 82%) as a yellow oil: MS (ESI): MH+ 389.
Intermediate 21 : /v"-(3-hvdroxypropyl)-1-(3-nitrophenyl)methanesulfonamide
To an ice-cold solution of 1-(3-nitrophenyl)-Λ/-{3-
[(triethylsilyl)oxy]propyl}methanesulfonamide (0.67g, 1.7mmol) in tetrahydrofuran (4 ml) was added TBAF (1 M in tetrahydrofuran, 3.4 ml) via syringe. The reaction mixture was stirred at ice bath temperature for 30 min, and then concentrated under reduced pressure. The resulting residue was partitioned between 3:1 chloroform/2- propanol (40ml) and water (20ml) and the aqueous layer extracted with 3:1 chloroform/2-propanol (30ml). The combined organic layers were dried (sodium sulfate) and then concentrated under reduced pressure. The crude product was purified by flash chromatography (silica, 95:5 methylene chloride/methanol) to afford Λ/-(3-hydroxypropyl)-1-(3-nitrophenyl)methanesulfonamide (0.37g, 79%) as a yellow oil: MS (ESI): MH+ 275.
Intermediate 22: 1 -(3-aminophenylV/V-(3-hvdroxypropyl)methanesulfonamide
A mixture of Λ/-(3-hydroxypropyl)-1-(3-nitrophenyl)methanesulfonamide (0.37g, 1.3mmol), and Pd/C (10%, 0.07g) in ethanol (30ml) were shaken under an atmosphere of hydrogen at 40 psi for 30min. The reaction mixture was filtered
through diatomaceous earth, washing with ethyl acetate (150ml). The filtrate was concentrated under reduced pressure to provide Λ/-(3-hydroxypropyl)-1-(3- aminophenyl)methanesulfonamide as a yellow oil: MS (ESI): MH+ 245.
Intermediate 23: 1-(3-aminopropyl)-2,5-pyrrolidinedione
A mixture of 3-(2,5-dioxo-1-pyrrolidinyl)propanenitrile (Lancaster, 2.Og, 13mmol), hydrochloric acid (12M, 2.7ml) and PtO2 (166mg, 0.70mmol) in ethanol (52ml) was hydrogenated at 50 psi for 16 h. The reaction mixture was filtered through diatomaceous earth. The filtrate was concentrated under reduced pressure to one third volume to afford a slurry which was filtered to provide 1-(3-aminopropyl)-2,5- pyrrolidinedione as a white solid which was used, without purification, in the next reaction.
Intermediate 24: Λ/-f3-(2,5-dioxo-1 -pyrrolidinyl)propyπ-2-(4-nitrophenyl)acetamide
To a solution of 1-(3-aminopropyl)-2,5-pyrrolidinedione (2.1g, 10.7mmol) in methylene chloride (63ml) at O0C under nitrogen were added 1-hydroxybenzotriazole (0.96g, 7.1mmol), para-nitrophenylacetic acid (Lancaster, 1.29g, 7.13mmol), 1-(3- dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (1.5Og, 7.84mmol), and Λ/,Λ/-diisopropy!ethylamine (9.3ml, 53mmol) respectively. The reaction mixture was allowed to warm to room temperature overnight, then diluted with methylene chloride (100ml), washed with 1 M hydrochloric acid (3 x 150ml) and a solution of saturated sodium bicarbonate (3 x 150ml). The organic layer was dried over anhydrous sodium sulfate, gravity filtered, and concentrated under reduced pressure to one-third volume to afford a slurry which was filtered to give Λ/-[3-(2,5-dioxo-1- pyrrolidinyl)propyl]-2-(4-nitrophenyl)acetamide as a yellow solid: MS (ESI): MH+ 320.
Intermediate 25: Λ/-f3-(2,5-dioxo-1 -pyrrolidinyl)propyπ-2-(4-aminophenyl)acetamide
A mixture of /V-[3-(2,5-dioxo-1-pyrrolidinyl)propyl]-2-(4-nitrophenyl)acetamide (1.28g, 4.01mmol), and Pd/C (10%, 169mg, 0.08mmol) in ethanol (16ml) was hydrogenated at 50 psi for 7h. The reaction mixture was filtered through diatomaceous earth, and the filter cake washed with ethanol (300 ml). The filtrate was concentrated under reduced pressure to provide A/-[3-(2,5-dioxo-1-pyrrolidinyl)propyl]-2-(4- aminophenyl)acetamide as an off-white solid: MS (ESI): MH+ 290
Intermediate 26: 2-(3-nitrophenyl)-Λ/-r3-(2-oxo-1 -pyrrolidinvDpropyllacetamide
To a solution of 1-(3-aminopropyl)-2-pyrrolidinone (Aldrich, 5.8g, 30mmol) in methylene chloride (69ml) at O0C were added 1-hydroxybenzotriazole (3.7g, 28mmol), meta-nitrophenylacetic acid (5.0Og, 28mmol), 1-(3-dimethylaminopropyl)-3- ethyl carbodiimide hydrochloride (5.8g, 30mmol), and /V,Λ/-diisopropylethylamine (28ml, 170mmol) respectively. The reaction mixture was was allowed to warm to room temperature overnight, then diluted with methylene chloride (100ml), washed
with hydrochloric acid (1N, 3 x 150ml) and a solution of saturated sodium bicarbonate (3 x 150ml). The organic layer was dried over anhydrous sodium sulfate, gravity filtered, and concentrated under reduced pressure to afford 2-(3-nitrophenyl)-Λ/-[3-(2- oxo-1-pyrrolidinyl)propyl]acetamide as a yellow oil which was used, without purification, in the next reaction.
Intermediate 27: 2-(3-aminophenyl)-A/-[3-(2-oxo-1-pyrrolidinyl)propyllacetamide
A mixture of 2-(3-nitrophenyl)-A/-[3-(2-oxo-1-pyrrolidinyl)propyl]acetamide (8.2g, 27mmol), and 10% Pd/C (1.72g, 0.81 mmol) in ethanol (54ml) was shaken under an atmosphere of hydrogen at 50 psi for 16h. The reaction mixture was filtered through diatomaceous earth, and the filter cake washed with ethanol (300ml). The filtrate was concentrated under reduced pressure and the crude product was purified over silica gel to afford 2-(3-aminophenyl)-Λ/-[3-(2-oxo-1-pyrrolidinyl)propyl]acetamide as an off-white solid: MS (ESI): MH+ 290. .
Intermediate 28: 2-(4-nitrophenyl)-Λ/-r3-(2-oxo-1 -pyrrolidinvDpropyllacetamide
To a solution of 1-(3-aminopropyl)-2-pyrrolidinone (Aldrich, 5.8g, 41 mmol) in methylene chloride (69ml) at O0C were added 1-hydroxybenzotriazole (3.7g, 28mmol), para-nitrophenylacetic acid (Lancaster, 5.Og, 28mmol), 1-(3- dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (5.8g, 30.4mmol), and N, N- diisopropylethylamine (28ml, 170mmol) respectively. The reaction mixture was allowed to warm to room temperature overnight, then diluted with methylene chloride (100ml), washed with 1 M hydrochloric acid (3 x 150ml) and a solution of saturated sodium bicarbonate (3 x 150ml). The organic layer was dried over anhydrous sodium sulfate, gravity filtered, and concentrated under reduced pressure to afford 2- (4-nitrophenyl)-Λ/-[3-(2-oxo-1-pyrrolidinyl)propyl]acetamide (4.5g, 52%) as an off- white solid: MS (ESI): MH+ 306.
Intermediate 29: 2-(4-aminophenyl)-Λ/-r3-(2-oxo-1 -pyrralidinvDpropyllacetamide
To a solution of 2-(4-nitrophenyl)-Λ/-[3-(2-oxo-1-pyrrolidinyl)propyl]acetamide (2.8g, 9.1 mmol) in ethanol (18ml) was added tin(ll)chloride (8.7g, 45.7mmol) and the solution was heated at 7O0C under nitrogen for 2h. The reaction mixture was concentrated under reduced pressure, diluted with ethyl acetate (150ml), and washed with a solution of 2M sodium hydroxide (2 x 200ml). The organic layer was dried over anhydrous sodium sulfate, gravity filtered, and concentrated under reduced pressure to afford 2-(4-aminophenyl)-Λ/-[3-(2-oxo-1~ pyrrolidinyl)propyl]acetamide (409mg, 17%) as an off-white solid: MS (ESI): MH+ 276.
Examples
The names of the following Examples have been obtained from the structures shown, using the compound naming programme "ACD Name Pro 6.02":
Experimental details for the preparation of representative Examples are given in full below. Summary details for Examples 2 to 84 which were prepared by analogous methods are given in the accompanying tables.
Example 1 : formic acid - 5-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyπamino)-1 ,3- dihvdro-2H-indol-2-one (1 :1)

A mixture of N-(2-chloro-4-pyrimidinyl)-1H-indazol-4-amine (40mg) and 5- aminooxindole (Combi-blocks and others, 30mg) in acetone:water:2M HCI (2ml_:3mL:40ul_) was stirred and heated under reflux for 16h. The solvent was evaporated under nitrogen blowdown. The residue was diluted in methanol and eluted through an acid ion exchange column (type SCX). The product was released from the column using 10% ammonia in methanol. After concentration under nitrogen blowdown, the product was purified by mass directed HPLC (I) to give the title compound (12mg). LC/MS: Rt 2.1min, MH+ 358.
Similarly prepared were the following examples 2 to 84, shown in table 1 below:
Table 1













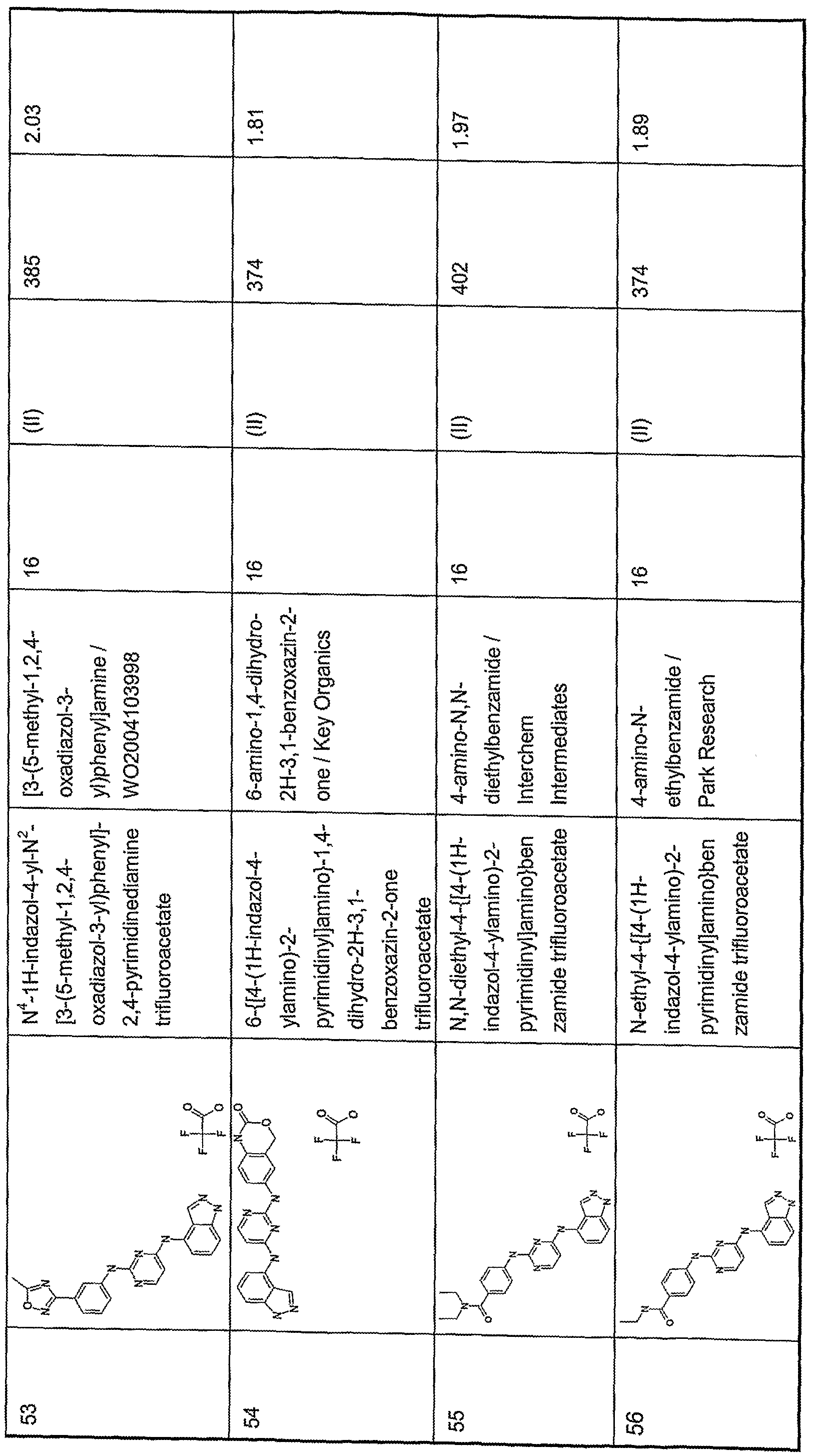










(a) Salt forms: HCI = hydrochloride; HCO2H = formate if no salt form is listed the compound is a free base
(b) Isolation Method: (I) Mass Directed HPLC Method A; compounds isolated by this method are formate salts or free bases.
(II) Mass Directed HPLC Method B; compounds isolated by this method are trifluoroacetate salts.
Examples 85- 140 : General purification and analytical methods
Analytical HPLC was conducted by five different methods:
Method A (default) - on a Supelcosil LCABZ+PLUS column (3um, 3.3cm x 4.6mm ID) eluting with 0.1% HCO2H and 0.01 M ammonium acetate in water (solvent A), and 95% acetonitrile and 0.05% HCO2H in water (solvent B), using the following elution gradient 0-0.7 minutes 0%B, 0.7-4.2 minutes 0→100%B, 4.2-5.3 minutes 100%B, 5.3-5.5 minutes 100→0%B, flow 3 ml/min.
Method B - on a Symmetry C18 column (250 x 4.6 mm, 5um), eluting with H2O with 0.05% TFA (solvent A), and CH3CN with 0.05% TFA (solvent B), using the following eluton gradient 10-90% B over 15 min; flow 1.0 mL/min. Detection: 230 or 254 nm.
Method C - on a Symmetry Prep C18 column (300 x 19 mm, 7um), eluting with H2O with NH4OAc/AcOH buffer (solvent A) and MeOH (solvent B), using the following elution gradient 20-100% B over 20 min; flow 17.0 mL/min. Detection: 230 nm.
Method D - on an Alltech Platinum Rocket C18 column (53 x 7 mm, 5um), eluting with H2O with 0.05% TFA (solvent A), and CH3CN with 0.05% TFA (solvent B), using the following gradient 10-90% B over 15 min; flow 1.0 mL/min. Detection: 230 nm
Method E - on an Alltech Platinum Rocket C18 column (53 x 7 mm, 5Dm), eluting with H2O with 0.05% TFA (solvent A) and CH3CN with 0.05% TFA (solvent B), using the following gradient 10-90% B over 15 min; flow 1.0 mL/min. Detection: 230 nm
Mass spectra (MS) were recorded on a Fisons VG Platform mass spectrometer using electrospray positive ionisation [(ES+ve to give MH+ and M(NH4)4" molecular ions] or electrospray negative ionisation [(ES-ve to give (M-H)" molecular ion] modes, or on a Perkin Elmer Sciex 100 atmospheric pressure ionization (APCI) mass spectrometer, or a Finnigan LCQ Duo LC/MS Ion Trap electrospray ionization (ESI) mass spectrometer. Retention time in LC/MS is referred to as "Rt" (time in minutes).
"Mass directed autoprep" refers to methods where the material was purified by high performance liquid chromatography on a HPLCABZ+ 5μm column (5cm x 10mm i.d.) with 0.1% HCO2H in water and 95% MeCN, 5% water (0.5% HCO2H) utilizing the following gradient elution conditions: 0-1.0 minutes 5%B, 1.0-8.0 minutes 5→30%B, 8.0-8.9 minutes 30%B, 8.9-9.0 minutes 30→95%B, 9.0-9.9 minutes 95%B, 9.9-10 minutes 95→0%B at a flow rate of 8ml minutes"1 (System 2). The Gilson 202-fraction collector was triggered by a VG Platform Mass Spectrometer on detecting the mass of interest.
"Biotage™ chromatography" refers to purification carried out using equipment sold by Dyax Corporation (either the Flash 4Oi or Flash 15Oi) and cartridges pre-packed with KPSiI.
Examples 85 to 104.
These were prepared according to the method:
A mixture of Λ/-(2-chloro-4-pyrimidinyl)-1/-/-indazol-4-amine (30.0 mg) and aniline (0.128 mmol) in acetone/water/conc. hydrochloric acid (150:100:2, 1.5 ml) was heated at 750C overnight. The reaction was then evaporated to dryness and the crude residue was purified by mass directed autoprep to give the desired product. These are shown in Table 2 below:
Table 2





To a solution of [(4-{[4-(1/-/-indazol-4-ylamino)-2-pyrimidinyl]amino}phenyl)oxy]acetic acid (0.3Og, 0.44mmol) and Λ/,Λ/-diisopropylethylamine (0.34g, 2.60mmol), in N, N- dimethylformamide (4ml) was added HBTU (0.2Og, 0.53mmol). The reaction mixture was stirred at room temperature for 15min before a solution of 3-aminopropanol (0.10g, 1.3mmol) in Λ/,Λ/-dimethylformamide (1.5ml) was added via syringe. After stirring for 2Oh, the reaction mixture was partitioned between water (50ml) and 3:1 chloroform/2-propanol (150ml). The organic layer was washed with water (50ml), a 5% solution of lithium chloride (50ml), and then dried (sodium sulfate) and concentrated under reduced pressure. The crude product was purified by flash chromatography (silica gel, 9:1 methylene chloride/methanol) to afford the title compound as an off-white solid: mp 94-98 0C; 1H NMR (500 MHz, DMSO-Cf6) δH 13.04 (s, 1 H), 9.34 (s, 1 H), 9.04 (s, 1 H), 8.29 (s, 1H), 8.07-8.04 (m, 2H), 7.92-7.91 (m, 1 H), 7.62 (d, J = 9.0 Hz, 2H), 7.27 (t, J = 8.0 Hz, 1 H), 7.18 (d, J = 8.2 Hz, 1 H), 6.86 (d, J = 9.0 Hz, 2H), 6.41 (d, J = 5.7 Hz, 1 H), 4.47 (t, J = 5.2 Hz, 1 H), 4.41 (s, 2H), 3.44-3.40 (m, 2H), 3.22-3.18 (m, 2H), 1.62-1.57 (m, 2H); MS (ESI): MH+ 434; HPLC (Method D): >99% (AUC), Rt 2.87min.
Example 106: Methyl Λ/-{[(4-(r4-(1H-indazol-4-ylamino)-2- pyrimidinyl1amino)phenyl)oxylacetyl) serinate

To a solution of [(4-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyl]amino}phenyl)oxy]acetic acid (0.27g, 0.40mmol) and Λ/,/V-diisopropylethylamine (0.2Og, 1.6mmol), in N,N- dimethylformamide (3.5ml) was added HBTU (0.18g, 0.48mmol). The reaction mixture was stirred at room temperature for 15min before a solution of methyl
serinate (Aldrich, 0.19g, 1.2mmol) and Λ/,Λ/-diisopropylethylamine (0.1Og, O.δmmol) in Λ/,Λ/-dimethylformamide (1.5ml) was added via syringe. After stirring for 15 h, the reaction mixture was diluted with ethyl acetate (100ml) and washed with water (3 x 50ml) and a 5% lithium chloride solution (50ml). The organic layer was dried (sodium sulfate), concentrated under reduced pressure, and the crude product was purified by flash chromatography (silica gel, 9:1 methylene chloride/methanol) to afford the title compound (as a mixture of enantiomers) as a pale yellow solid: mp 115-117 °C; 1H NMR (500 MHz, DMSO-c/6) δH 13.03 (s, 1 H), 9.34 (s, 1 H), 9.05 (s, 1 H), 8.29 (s, 1H), 8.18 (d, J = 7.8 Hz, 1H), 8.05 (d, J = 5.7 Hz, 1 H), 7.92-7.91 (m, 1 H), 7.63 (d, J = 9.0 Hz, 2H), 7.30-7.26 (m, 1H), 7.18 (d, J = 8.2 Hz, 1H), 6.87 (dd, J = 7.0, 2.0 Hz, 2H), 6.40 (d, J = 5.7 Hz, 1 H), 5.16 (t, J = 5.7 Hz, 1 H), 4.52 (d, J = 2.2 Hz, 2H), 4.47-4.44 (m, 1 H), 3.81-3.76 (m, 1H), 3.71-3.67 (m, 1 H), 3.65 (s, 3H); MS (ESI): MH+ 478; HPLC (Method B): 96.0% (AUC), Rt 8.30min.
Example 107: Λ/-(3-hvdroxypropyl)-1-(3-(r4-(1H-indazol-4-ylamino)-2- pyrimidinyl1amino}phenyl)methanesulfonamide

To a mixture of 1-(3-aminophenyl)-Λ/-(3-hydroxypropyl)methanesulfonamide (0.16g, 0.66mmol) and 1 ,1-dimethylethyl 4-[(2-chloro-4-pyrimidinyl)amino]-1H-indazole-1- carboxylate (0.2Og, 0. 59mmol) in 2-propanol (5ml) at 800C was added 2M hydrochloric acid in diethyl ether (1 drop). The resulting mixture was stirred at 800C, under nitrogen, for two days. The reaction mixture was concentrated under reduced pressure, the residue diluted with methylene chloride (5ml), and trifluoroacetic acid (1 ml) was added. After stirring at room temperature for 30min, the reaction mixture was diluted with methylene chloride (50ml), and carefully neutralized by saturated sodium bicarbonate solution. The solid which had formed was collected by vacuum filtration, dissolved in methanol and the combined organic solvents were concentrated under reduced pressure. The crude product was purified by flash chromatography (silica gel, 9:1 methylene chloride/methanol) was to afford the title compound as a pale yellow solid: mp 127-132 0C; 1H NMR (500 MHz, DMSO-d6) δH 13.05 (s, 1 H), 9.39 (s, 1H), 9.28 (s, 1 H), 8.29 (s, 1 H), 8.09 (d, J = 5.7 Hz, 1H), 7.94-7.92 (m, 1 H), 7.75-7.74 (m, 2H), 7.31-7.28 (m, 1 H), 7.23-7.19 (m, 2H), 6.97 (t, J = 5.7 Hz, 1 H), 6.91 (d, J = 7.5 Hz, 1 H), 6.46 (d, J = 5.7 Hz, 1 H), 4.40 (t, J = 5.0 Hz,
1H), 4.17 (s, 2H), 3.38 (dd, J = 11.4, 6.1 Hz, 2H), 2.93-2.89 (m, 2H), 1.56-1.51 (m, 2H); MS (ESI): MH+ 454; HPLC: (Method D) >99% (AUC), Rt 2.90min.
Example 108: Λ/-[3-(2,5-dioxo-1 -pyrrolidinyl)propyπ-2-(4-(r4-(1 /-/-indazol-4-ylaminoV2- pyrimidinyllamino) phenvQacetamide

To a mixture of Λ/-[3-(2,5-dioxo-1-pyrrolidinyl)propyl]-2-(4-aminophenyl)acetamide (138mg, 0.48mmol) and 1 ,1-dimethylethyl 4-[(2-chloro-4-pyrimidinyl)amino]-1H- indazole-1-carboxylate (150mg, 0.43mmol) in 2-propanol (3ml) at 800C was added 2M hydrochloric acid in diethyl ether (1 drop). The resulting mixture was stirred at 8O0C- under nitrogen for 16h. The reaction mixture was concentrated under reduced pressure, the residue diluted with methylene chloride (6ml), and trifluoroacetic acid (1ml) was added. After stirring at room temperature for 30min, the reaction mixture was agitated with a solution of saturated sodium bicarbonate (100ml). The resulting slurry was filtered and the crude product was purified over silica gel to afford the title compound.as an off-white solid: mp 120-123 °C; 1H NMR (500 MHz, DMSO-Cf6) δH 13.05 (s, 1 H)1 9.35 (s, 1 H), 9.13 (s, 1 H), 8.29 (s, 1 H), 8.07 (d, J = 5.7 Hz, 1 H), 7.93 (d, J = 6.1 Hz, 2H), 7.64 (d, J = 8.6 Hz1 2H), 7.26 (d, J = 7.9 Hz, 1H), 7.19 (d, J = 8.3 Hz, 1 H)1 7.10 (d, J = 4.5 Hz, 2H), 6.43 (d, J = 5.8 Hz, 1H), 3.29-3.25 (m, 4H), 3.02 (d, J = 6.1 Hz, 2H), 2.59 (s, 4H), 1.61-1.59 (m, 2H); MS (ESI): MH+ 499, HPLC (Method B) >99%: Rt 9.6 min.
Example 109: 2-(3-(f4-(1 /-/-indazol-4-ylamino)-2-pyrimidinyllamino)phenyl)-A/-r3-(2- oxo-1-pyrrolidinyl)propyllacetamide

To a mixture of 2-(3-aminophenyl)-A/-[3-(2-oxo-1-pyrrolidinyl)propyl]acetamide (92mg, 0.31 mmol) and 1 ,1-dimethylethyl 4-[(2-chloro-4-pyrimidinyl)amino]-1H~ indazole-1-carboxylate (100mg, 0.3 mmol) in 2-propanol (3ml) at 80 0C was added
2M hydrochloric acid in diethyl ether (1 drop). The resulting mixture was stirred at 80 0C under nitrogen for 16 h. The reaction mixture was concentrated under reduced pressure, the residue diluted with methylene chloride (6ml), and trifluoroacetic acid (1 ml) was added. After stirring at room temperature for 30min, the reaction mixture was agitated with a solution of saturated sodium bicarbonate (100ml). The resulting slurry was filtered and the crude product was purified over silica gel to afford the title compound as an off-white solid: mp 130-135 °C; 1H NMR (500 MHz, DMSO-d6) δH 13.04 (s, 1H), 9.35 (s, 1H), 9.16 (s, 1H), 8.31 (s, 1H), 8.08 (d, J = 5.7 Hz, 1 H), 7.95- 7.94 (m, 2H), 7.63 (d, J = 8.2 Hz, 1 H), 7.60 (s, 1H), 7.28 (t, J = 8.0 Hz, 1H), 7.18 (d, J = 8.3 Hz, 1H), 7.14 (t, J = 7.8 Hz, 1 H), 6.82 (d, J = 7.6 Hz, 1 H), 6.45 (d, J = 5.7 Hz, 1 H), 3.33 (s, 2H), 3.27 (t, J = 4.9 Hz, 2H), 3.15 (t, J = 7.1 Hz, 2H), 3.03-2.99 (m, 2H), 2.18 (t, J = 8.0 Hz, 2H), 1.88 (t, J = 7.5 Hz, 2H), 1.57 (t, J = 7.1 Hz, 2H); MS (ESI): MH+ 485; HPLC (Method B): 97.1%, Rt 8.3min.
Example 110: 1 ,1 -dimethylethylf(4-{f4-(1 H-indazol-4-ylamino)-2-pyrimidinvπamino) phenyQoxylacetate

Under nitrogen Λ/-(2-chloro-4-pyrimidinyl)-1H-indazol-4-amine (200mg, 0.814mmol) and 1 ,1-dimethylethyl [(4-aminophenyl)oxy]acetate (Chemical & Pharmaceutical Bulletin (1999), 47(12), 1685-1693, 360mg, 1.63mmol) were dissolved in isopropanol (5ml and was heated to 84°C for 16h. The reaction was then diluted with 3:1 CHCl3/2-PrOH (100ml) and washed with NaHCC>3. The organic layer was dried over Na2SC>4 and concentrated to give a brown oil which was purified by flash chromatography to yield the title compound. MS (ESI): MH+ 433.
Example 111 : 1-(4-{r4-(1H-indazol-4-ylamino)-2-pyrimidinyllamino)phenvn-/V- methylmethanesulfonamide

The title compound was prepared following the procedure of Example 110 with N-(2- chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 1 -(4-aminophenyl)-A/- methylmethanesulfonamide (Lancaster). MS (ESI): MH+ 410.
Example 112: 2-IY4-(r4-(1/-/-indazol-4-ylaminoV-2- pyrimidinyliaminolphenyPoxyiacetamide
The titled compound was prepared following the procedure of Example 110 with N- (2-chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 2-[(4-aminophenyl)oxy]acetamide (AKos Screening Library). MS (ESI): MH+ 376.
Example 113: 2-K4-(r4-(1H-indazol-4-ylamino)-2-pyrimidinyriamino)phenyl)oxy1-Λ/- methylacetamide

The title compound was prepared following the procedure of Example 110 with N-(2- chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 2-[(4-aminophenyl)oxy]-Λ/- methylacetamide (WO 951595). MS (ESI): MH+ 390.
Example 114: r(4-{f4-(1/-/-indazol-4-ylamino)-2-pyrimidinyllamino)phenyl)oxylacetic acid hydrochloride

Cl
The title compound was prepared following the procedure of Example 110 with N-(2- chloro-4-pyrimidinyl)-1 H-indazol-4-amine and [(4-aminophenyl)oxy]acetic acid (Interchim Intermediates). MS (ESI): MH+ 377.
Example 115: Methyl r(4-{[4-(1/-/-indazol-4-ylamino)-2-pyrimidinyllamino)phenyl)oxy1 acetate

The title compound was prepared following the procedure of Example 110 with N-(2- chloro-4-pyrimidinyl)-1 H-indazol-4-amine and methyl [(4-aminophenyl)oxy]acetate (Scientific Exchange Product List). MS (ESI): MH+ 390.
Example 116: Λ/-(3,4-dimethyl-5-isoxazolyl)-1-(4-{f4-(1H-indazol-4-ylamino)-2- pyrimidinyllamino)phenyl)methanesulfonamide

The titled compound was prepared following the procedure of Example 110 with N- (2-chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 1-(4-aminophenyl)-Λ/-(3,4-dimethyl- 5-isoxazolyl)methanesulfonamide. MS (ESI): MH+ 491.
Example 117: Λ/-(4,5-dimethyl-1.3-thiazol-2-ylV1-f4-ff4-(1 Wndazol-4-ylamino)-2- pyrimidinvnamino}phenyl)methanesulfonamide

The title compound was prepared following the procedure of Example 110 with N-(2- chloro-4-pyrimidinyl)-1H-indazol-4-amine and 1-(4-aminophenyl)-/V-(4,5-dimethyl-1 ,3- thiazol-2-yl)methanesulfonamide. MS (ESI): MH+ 507.
Example 118: 1-(4-{f4-(1f/-indazol-4-ylamino)-2-pyrimidinyllamino)phenyl)-A/-f3-(2- oxo-1-pyrrolidinyl)propyllmethanesulfonamide

The title compound was prepared following the procedure of Example 110 with N-(2- chloro-4-pyrimidinyl)-1H-indazol-4-amine and 1-(4-aminophenyl)-Λ/-[3-(2-oxo-1- pyrrolidinyl)propyl]methanesulfonamide. MS (ESI): MH+ 521.
Example 119: Λ/-(3-hvdroxypropyl)-1-(4-{f4-(1/-/-indazol-4-ylamino)-2- pyrimidinyllamino}phenyl)methanesulfonamide

1 , 1 -Dimethylethyl 4-[(2-chloro-4-pyrimidinyl)amino]-1 /-/-indazole-1 -carboxylate
(0.569mmol) and 1 -(4-aminophenyl)-Λ/-(3-hydroxypropyl)methanesulfonamide (0.626mmol) were dissolved in 2-PrOH (5ml) with a few drops of 2M HCI in ether.
The reaction was heated at 80°C under N2 for 3 days. The reaction was concentrated in vacuo and the residue was dissolved in methylene chloride (6ml). After TFA (1ml) was added, the reaction was stirred for 30min. The reaction was
diluted with methylene chloride (100ml) and carefully neutralized with saturated NaHCθ3. The organic layer was then dried over IS^SO^ filtered and concentrated to give a solid. The solid was purified by flash chromatography to afford the title compound. MS (ESI): MH+ 454.
Example 120: 2-[(4-([4-(I /-/-indazol-4-ylamino)-2-pyrimidinyllamino}phenyl)oxy1-A/-r3- (2-oxo-1-pyrrolidinyl)propyllacetamide

To a solution of [(4-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyl]amino}phenyl)oxy]acetic acid (300mg, 0.439mmol) and DIPEA (0.46ml) in DMF (4ml) was added HBTU (200mg, 0.527mmol). The mixture was stirred at room temperature under nitrogen for 15min before a solution of 1-(3-aminopropyl)-2-pyrrolidinone (Aldrich, 190mg, 1.32mmol) in DMF (2ml) was added. The reaction was stirred at room temperature under N2 overnight. The reaction was then diluted with water 50ml and extracted with
EtOAc. The combined organic layers were washed with water, dried over sodium sulphate^ filtered and concentrated to give a solid. The solid was purified by recrystallization in EtOAc to yield the titled compound. MS (ESl): MH+ 501.
Example 121 : Λ/-(3,4-dimethyl-5-isoxazolyl)-2-(4-ff4-(1 A7-indazol-4-ylamino)-2- pyrimidinvπaminolphenvPacetamide
N-(2-chloro-4-pyrimidinyl)-1 H-indazol-4-amine (280mg, 1.1mmol) and 2-(4- aminophenyl)-/v-(3,4-dimethyl-5-isoxazolyl)acetamide (280mg, 1.1 mmol) were dissolved in IPA (7 ml) with a few drops of 2N HCI in ether. The reaction was heated for 3 days and then concentrated in vacuo. The mixture was dissolved in methylene chloride (5ml) treated with TFA (3ml) and stirred at room temperature for 1h and then quenched with NaHCθ3. The resulting solid was filtered and purified by flash chromatography to yield the title compound. MS (ESl): MH+ 455'
Example 122: 2-(4-IK-(I H-indazol-4-ylaminoV2-pyrimidinyriamino}phenvO-Λ/-(5- methyl-3-isoxazolvPacetamide

The titled compound was prepared following the procedure of Example 121 with 1 ,1- Dimethylethyl 4-[(2-chloro-4-pyrimidinyl)amino]-1/-/-indazole-1-carboxylate and 2-(4- aminophenyl)-A/-(5-methyl-3-isoxazolyl)acetamide. MS (ESI): MH+ 441.
Example 123: N-{4-r(difluoromethv0oxylphenyl)-2-(4-{l4-(1 /-/-indazol-4-ylamino)-2- Pyrimidinyllamino)phenvOacetaιτiide

N-(2-chloro-4-pyrimidinyl)-1 H-indazol-4-amine (1.02mmol) 2-(4-aminophenyl)-Λ/-{4- [(difluoromethyl)oxy]phenyl}acetamide (1.13mmol) were dissolved in IPA (7ml) with a few drops of 2N HCI in EtOH. The reaction was heated at reflux for 3 days and then concentrated in vacuo and dissolved in methylene chloride (5ml) and treated with TFA (5ml). The reaction was stirred at room temperature for 1 h and then quenched with NaHCθ3- The resulting solid was filtered and purified by flash chromatography to yield the title compound. MS (ESI): MH+ 502.
Example 124: Λ/M H-indazol-4-yl-Λ/2-(4-r(methylsulfonyl)methyllphenyl>-2,4- pyrimidinediamine

The title compound was prepared following the procedure of Example 123with N-(2- chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 4-[(methylsulfonyl)methyl]aniline (Bioorganic & Medicinal Chemistry Letters (2004), 14(1 ), 187-190). MS (ESI): MH+ 395.
Example 125: 2-(4-(K-(I H-indazol-4-ylamino)-2-pyrimidinyllamino|phenvQ-Λ/-r4- (methyloxy)phenyllacetamide

The titled compound was prepared following the procedure of Example 123_with N- (2-chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 2-(4-aminophenyl)-Λ/-[4- (methyloxy)phenyl]acetamide (US 2004220247). MS (ESI): MH+ 466.
Example 126: 2-(4-{[4-(1 H-indazol-4-ylamino)-2-pyrimidinyllamino)phenvO-Λ/-r3- (methyloxy)phenyllacetamide

The titled compound was prepared following the procedure of Example 123_with N- (2-chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 2-(4-aminophenyl)-Λ/-[3- (methyloxy)phenyl]acetamide. MS (ESI): MH+ 466.
Example 127: 4-{r4-(1/-/-indazol-4-ylaminoV2-pyrimidinyl1amino)benzoic acid

Methyl 4-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyl]amino}benzoate (1.67mmol) was dissolved in THF/MeOH/2M NaOH (3:1:1, 6ml) and stirred at room temperature for overnight. The reaction was concentrated in vacuo and water (10ml) added. The
mixture was acidified with 1 M HCI and the resulting solid was filtered off and purified by HPLC to yield the title compound as an off white solid. MS (ESI): MH+ 347.
Example 128: Methyl 4-{r4-(1H-indazol-4-ylamino)-2-pyrimidinvπamino)benzoate

N-(2-Chloro-4-pyrimidinyl)-1H-indazol-4-amine (0.87mmol), 3-amino benzoic acid methyl ester (Fluka, 1.22mmol), Pd2(dba)3 (10mg, 0.017mmol), K3PO4 (260mg,
1.218mmol) and Xantphos (30mg, 0.051 mmol) were dissolved in dioxane (5ml) under nitrogen. The reaction was stirred at 100°C for 18hrs and then diluted with THF. The resulting solid was filtered off and the filtrate was concentrated in vacuo to yield a yellow solid that was purified by flash chromatography to afford the title compound. MS (ESI): MH+ 361.
Example 129: 2-(4-(F4-(1 f/-indazol-4-ylamino)-2-pyrimidinvnamino}phenyl)-Λ/-f4- (trifluoromethyl)phenyllacetamide

The title compound was prepared following the procedure of Example 123 with N-(2- chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 2-(4-aminophenyl)-Λ/-[4- (trifluoromethyl)phenyl]acetamide. MS (ESI): MH+ 504.
Example 130: 2-(4-<T4-(1 H-indazol-4-ylamino)-2-pyrimidinyl1amino)phenyl)-Λ/-r3- (trifluoromethyl)phenvπacetamide

Example 131 : Λ/-(3-chlorophenvO-2-(4-(r4-(1 /-/-indazol-4-ylamino)-2- pyrimidinyliaminolphenyPacetamide

The title compound was prepared following the procedure of Example 123 with N-(2- chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 2-(4-aminophenyl)-Λ/-(3- chlorophenyl)acetamide. MS (ESI): MH+ 470.
Example 132: Λ/-(4-chlorophenvP-2-(4-(r4-(1 H-indazol-4-ylamino)-2- pyrimidinvnamino)phenyl)acetamide
The titled compound was prepared following the procedure of Example 123 with N- (2-chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 2-(4-aminophenyl)-Λ/-(4- chlorophenyl)acetamide (Journal of the American Chemical Society (1992), 114(9), 3528-34). MS (ESI): MH+ 470.
Example 133: Λ/-(4,5-dimethyl-1.3-thiazol-2-ylV2-(4-{f4-(1H-indazol-4-ylamino)-2- pyrimidinvπamino}phenyl)acetamide

The title compound was prepared following the procedure of Example 123 with Boc protected N-(2-chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 2-(4-aminophenyl)-Λ/- (4,5-dimethyl-1 ,3-thiazol-2-yl)acetamide. MS (ESI): MH+ 471.
Example 134: 2-(4-{f4-f1 H-indazol-4-ylamino)-2-pyrimidinyllamino)phenyl)-Λ/-r3-(2- oxo-1-pvrrolidinyl)propvllacetamide

The title compound was prepared following the procedure of Example 123 with N-(2- chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 2-(4-aminophenyl)-Λ/-[3-(2-oxo-1- pyrrolidinyl)propyl]acetamide MS (ESI): MH+ 485.
Example 135: 2-(4-{|4-(1 H-indazol-4-ylamino)-2-pyrimidinvπamino)phenvQ-Λ/-4- pyridinylacetamide

Λ/-(2-chloro-4-pyrimidinyl)-1W-indazol-4-amine (0.497mmol) and 2-(4-aminophenyl)- Λ/-4-pyridinylacetamide (0.547mmol) were dissolved in IPA (3ml). The reaction was heated at reflux for 3 days and then diluted with ether. The resulting solid was filtered off and purified by HPLC to yield the title compound MS (ESI): MH+ 455
Example 136: Λ/-(4-(r4-(1/-/-indazol-4-ylamino)-2- pyrimidinyπamino)phenyl)methanesulfonamide

The title compound was prepared following the procedure of Example 135 with N-(2- chloro-4-pyrimidinyl)-1 H-indazol-4-amine and Λ/-(4~ aminophenyl)methanesulfonamide (Interchim Intermediates). MS (ESI): MH+ 396.
Example 137: 2-(4-{f4-(1 Wndazol-4-ylamino)-2-pyrimidinynamino)phenyl)-/V- methylacetamide

The title compound was prepared following the procedure of Example 135 with N-(2- chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 2-(4~aminophenyl)-Λ/-methylacetamide (Journal of Pharmaceutical Sciences (1987), 76(1), 18-20). MS (ESI): MH+ 374.
Example 138: 2-(4-{r4-(1H-indazol-4-ylamino)-2-pyrimidinyl1amino)phenyl)-Λ/-3- pyridinylacetamide

The title compound was prepared following the procedure of Example 135 with N-(2- chloro-4-pyrimidinyl)-1 H-indazol-4-amine and 2-(4-aminophenyl)-Λ/-3- pyridinylacetamide. MS (ESI): MH+ 437.
Example 139: Λ/-(4-{[4-(1 H-indazol-4-ylamino)-2-pyrimidinvπamino!phenyl)-4- methylbenzenesulfonamide

The title compound was prepared following the procedure of Example 123_with 1,1- dimethylethyl 4-[(2-chloro-4-pyrimidinyl)amino]-1H~indazole-1-carboxylate and Λ/-(4- aminophenyl)-4-methylbenzenesulfonamide (Aldrich). MS (ESI): MH+ 472.
Example 140: Λ/M H-indazol-4-yl-Λ/2-r4-(1 H-tetrazol-5-ylmethvnphenvn-2.4- pyrimidinediamine

The title compound was prepared following the procedure of Example 123_with 1 ,1- dimethylethyl 4-[(2-chloro-4-pyrimidinyl)amino]-1H-indazole-1-carboxylate and [4- (1/-/-tetrazol-5-ylmethyl)phenyl]amine (Journal of Medicinal Chemistry (1992), 35(5), 944-53). MS (ESI): MH+ 385.
Claims
1. A compound of formula (I):

in which:
R1, R2 and R3 is each independently selected from hydrogen, halogen, hydroxy, -C1-6 alkyl, -NR5R6, -C,.6alkylene-NR5R6, -CN, -Co^ alkylene-CC^H, C(O)C1-6 alkoxy, -C1-2 alkyl substituted by 1 or more fluorine atoms), -C1-6 alkoxy, -C(O)C1-6alkyl, - C(O)NR5R6, -C(O)NHR7, -OCH2C(O)NHR8, -OCH2C(O)OR5, -CH2C(O)NHR8, -NR5C(O)R6, -SC1-6alkyl, -S(O)C1-6alkyl, -S(O)2C1-6alkyl, -CH2S(O)2C1-6alkyl, - NHS(O)2R9, -S(O)3H, -S(O)2NR5R6, -S(O)2NR5R10, or -CH2S(O)2NR5R10, such that at least one of R1, R2 and R3 is hydrogen; or
R1 and R3 is each hydrogen and R2 is C3-7cycloalkyl, benzyloxy, benzimidazol-2yl, a 5- or 6-membered heteroaryl ring or a heterocyclic ring comprising from 1 to 3 heteroatoms selected from O, N and S bonded to the phenyl ring through a ring carbon or nitrogen (if present) atom and which heterocyclic ring is unsubstituted or substituted on one or more ring carbon atoms by C1-6alkyl or oxo (=0), or on a ring nitrogen atom by C1-6alkyl; or
R1 is hydrogen, halogen or C1-6 alkyl and R2 and R3 are attached to adjacent carbon atoms of the phenyl ring and are joined and together to form, in combination with the carbon atoms on the phenyl group to which they are attached, a 5- or 6-membered heteroaryl or heterocyclic ring ; or
R1 is hydrogen, halogen or C1-6 alkyl and R2 and R3 are attached to adjacent carbon atoms of the phenyl ring and are joined together to form, in combination with the carbon atoms on the phenyl group to which they are attached, a 5- or 6-membered carbocyclic ring, and
R5 and R6 are is each independently hydrogen or C1-6alkyl, or R5 and R6 together with the nitrogen atom to which they are attached form a 5- or 6-membered heterocyclic ring which may comprise a second heteroatom selected from O, S or N and which may be substituted on the second N, if present, by Ci-6alkyl , and which may be substituted on a ring carbon by oxo (=0), C1-6alkyl, di- C1-6alkyl (which may be the same or different), or halogen ; R7 is phenyl optionally substituted by halogen, C1-6alkyl;
R8 is hydrogen, Ci-6alkyl optionally terminally substituted with hydroxy, 2-oxo-1- pyrrolidinyl or 2,5-dioxo-i-pyrrolidinyl, a 5- or 6-membered heteroaryl ring comprising 1 or 2 heteroatoms independently selected from O, N and S and optionally substituted on a ring carbon by C1-6alkyl, phenyl optionally substituted by halo, C-rβalkyl, C1-6alkoxy or trifluoromethyl, or, together with the nitrogen to which it is attached is a C1-6alkyl ester of serine ;
R9 is Ci-6 alkyl or phenyl optionally substituted by Ci-6alkyl;
R10 is C1-6alkoxy, C1-6alkyl optionally terminally substituted with hydroxyl, 2-oxo-1- pyrrolidinyl or 2,5-dioxo-1-pyrrolidinyl, or R10 is a 5 or 6-membered heteroaryl ring comprising 1 heteroatom selected from O, N and S or 2 different heteroatoms independently selected from O, N and S and optionally substituted on one or more ring carbons by C1-6alkyl;
a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
2. A compound as claimed in claim 1 which is of the formula (Ia):

in which:
R1, R2a and R3a is each independently selected from the group of substituents Group A which comprises hydrogen, halogen, hydroxy, -C1-6 alkyl, -NR5R6, -Ci-6 alkylene-NR5R6, -CN, -C0-3alkylene-CO2H, C(O)C1-6 alkoxy, -C1-2 alkyl substituted by 1 or more fluorine atoms, -C1-6 alkoxy, -C(O)C1-6alkyl, -C(O)NR5R6, -C(O)NHR7, -OCH2C(O)NHR8, -OCH2C(O)OR5, -CH2C(O)NHR8, -NR5C(O)R6, -SC1- βalkyl. -S(O)C1-6alkyl, -S(O)2C1-6alkyl, -CH2S(O)2C1-6alkyl, -NHS(O)2R9, -S(O)3H1 -S(O)2NR5R6, -S(O)2NR5R10, or -CH2S(O)2NR5R10, such that at least one of R1, R2 and R3 is hydrogen; or
R1 is hydrogen and one of R2a and R3a is hydrogen and the other is R4 wherein R4 is benzyloxy, benzimidazol-2yl, a 5- or 6-membered heteroaryl ring or a heterocyclic ring comprising from 1 to 3 heteroatoms selected from O, N and S bonded to the phenyl ring through a ring carbon or nitrogen (if present) atom and which heterocyclic ring is unsubstituted or substituted on one or more ring carbon atoms by Ci-6alkyl or oxo (=0), or on a ring nitrogen atom by C1-6alkyl; or
R1 is hydrogen, halogen or C1-6 alkyl and R2a and R3a are joined and together form, in combination with the carbon atoms on the phenyl group to which they are attached, a 5- or 6-membered ring wherein: the ring is heteroaryl or heterocyclic and comprises one heteroatom selected from O, N or S, the ring is heteroaryl or heterocyclic and comprises two heteroatoms selected from O, N or S, which maybe the same or different, and excluding two S atoms, the ring is heteroaryl and comprises three heteroatoms of which two are N atoms and the third is nitrogen or oxygen, which ring may be unsubstituted or substituted on: one or more ring carbon atoms which each may be independently substituted by oxo (=O) or by 1 or 2 substituents which may be the same or different selected from C1-6 alkyl, hydroxy], or halo, one or more ring nitrogen atoms by C1-6 alkyl or C1-6 alkylcarbonyl, and a ring sulphur atom by (O)2; or
R1 is hydrogen, halogen or C1-6 alkyl and R2a and R3a are joined and together form, in combination with the carbon atoms on the phenyl group to which they are attached, a 5- or 6- membered non-aromatic carbocyclic ring which may be unsubstituted or substituted by oxo (=0), and
R5 and R6 are is each independently hydrogen or C1-6alkyl;
R7, R8, R9 and R10 are as hereinbefore defined for formula (I); or
a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
3. A compound as claimed in claim 1 or 2 in which two of R1, R2 and R3 are hydrogen.
4. A compound as claimed in claim 3 in which R1 and R3 is each hydrogen and R2 is benzyloxy, benzimidazol-2yl, a 5- or 6-membered heteroaryl ring or a heterocyclic ring comprising from 1 to 3 heteroatoms selected from O, N and S bonded to the phenyl ring through a ring carbon or nitrogen (if present) atom and which heterocyclic ring is unsubstituted or substituted on one or more ring carbon atoms by Ci-6alkyl or oxo (=O), or on a ring nitrogen atom by C1-6alkyl.
5. A compound as claimed in claim 4 in which R2 is selected from: benzimidazol-2-yl, tetrazol-5-yl ; 
6. A compound as claimed in claim 2 in which the 5- or 6- membered saturated or unsaturated ring formed by R2a and R3a, fused with the phenyl ring, is selected from:


7. A compound as claimed in claim 2 in which the 5- or 6- membered saturated carbocyclic ring formed by R2a and R3a include:

8. A compound as claimed in claim 1 selected from the compounds described as Examples 1 to 140 or salt or solvate thereof, in particular, a pharmaceutically acceptable salt or solvate thereof.
9. A compound as claimed in claim 1 selected from:#
6-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyl]amino}-3,4-dihydro-1(2H)-naphthalenone; N2-[3,4-bis(methyloxy)phenyl]-N4-1 H-indazol-4-yl-2,4-pyrimidinediamine; 5-{[4-(1 H- indazol-4-ylamino)-2-pyrimidinyl]amino}-1 ,3-dimethyl-1 ,3-dihydro-2H-benzimidazol-2- one;
N-(3-{[4-(1 H-indazol-4-ylamino)-2-pyrimidinyl]amino}phenyl)methanesulfonamide; N2-[3,5-bis(methyloxy)phenyl]-N4-1 H-indazol-4-yl-2,4-pyrimidinediamine; N4-1 H-indazoI-4-yl-N2-(1 -methyl-1 H-benzimidazol-5-yl)-2,4-pyrimidinediamine;
N4-1 H-indazol-4-yl-N2-(1 -methyl-1 H-indazol-5-yl)-2,4-pyrimidinediamine;
N4-1H-indazol-4-yl-N2-[3-(methylsulfonyl)phenyl]-2,4-pyrimidinediamine;
N^CS-chloro^-CmethyloxyjphenylJ-N^I H-indazol^-yl^^-pyrimidinediamine;
2-[(3-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyl]amino}phenyl)oxy]-N-methylacetamide;
6-{[4-(1 H-indazol-4-ylamino)-2-pyrimidinyl]amino}-2-benzofuran-1(3H)-one;
N4-1 H-indazol-4-yl-N2-[3-(1 ,3-oxazol-5-yl)phenyl]-2,4-pyrimidinediamine;
N4-1 H-indazol-4-yl-N2-[3-(5-methyl-1 ,2,4-oxadiazol-3-yl)phenyl]-2,4- pyrimidinediamine;
N-ethyl-4-{[4-(1 H-indazol-4-ylamino)-2-pyrimidinyl]amino}benzamide;
N2-1 ,3-benzodioxol-5-yl-N4-1 H-indazol~4-yl-2,4-pyrimidinediamine; N4-1 H-indazol-4- yl-N2-(1 -methyl-1 H-indazol-6-yl)-2,4-pyrimidinediamine;
N4-1H-indazol-4-yl-N2-[3-(1H-pyrazol-3-yl)phenyl]-2,4-pyrimidinediamine;
N4-1 H-indazol-4-yl-N2-1 H-indazol-5-yl-2,4-pyrimidinediamine;
N4-1 H-indazol-4-yl-N2-[3-(methyloxy)phenyl]-2,4-pyrimidinediamine;
3-{[4-(1 H-indazol-4-ylamino)-2-pyrimidinyl]amino}benzonitrile;
N4-1H-indazol-4-yl-N2-[3-(trifluoromethyl)phenyl]-2,4-pyrimidinediamine;
N4-1H-indazol-4-yl-N2-[3-(4-methyl-1,3-oxazol-5-yl)phenyl]-2,4-pyrimidinediamine;
3-{[4-(1 H-indazol-4-ylamino)-2-pyrimidinyl]amino}phenol; and methyl 3-{[4-(1H-indazol-4-ylamino)-2-pyrimidinyl]amino}benzoate; or salt or solvate thereof, in particular, a pharmaceutically acceptable salt or solvate thereof.
10. A process for preparing a compound of formula (I), or a salt or solvate thereof, as defined in claim 1 , which process comprises:
(a) reacting a compound of formula (II):

or a protected derivative thereof, wherein R1, R2, and R3 are as defined above and L1 represents a suitable leaving group, with a compound of formula (III)

or a protected derivative thereof; or (b) reacting a compound of formula (IV):

or a protected derivative thereof wherein L2 represents a suitable leaving group, with a compound of formula (V)

or a protected derivative thereof wherein R , R and R are as defined above.
11. A pharmaceutical formulation comprising a compound of formula (I), or a salt or solvate thereof, as defined in claim 1 and pharmaceutically acceptable excipients.
12. A compound of formula (I) as defined in claim 1 or a salt or solvate thereof for use in therapy.
13. The use of a compound of formula (I) as defined in claim 1 or a salt or solvate thereof in the manufacture of a medicament for treating a disease associated with inappropriate mast cell activation
14. The use of a compound of formula (I) as defined in claim 1 or a salt or solvate thereof in the manufacture of a medicament to inhibit a Syk kinase.
15. The use of a compound of formula (I) as defined in claim 1 or a salt or solvate thereof in the manufacture of a medicament for of treating an inflammatory disease
16. The use of a compound of formula (I) as defined in claim 1 or a salt or solvate thereof in the manufacture of a medicament for treating an allergic disorder.
17. The use of a compound of formula (I) as defined in claim 1 or a salt or solvate thereof in the manufacture of a medicament for treating rhinitis.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0511391A GB0511391D0 (en) | 2005-06-03 | 2005-06-03 | Novel compounds |
| GB0511391.5 | 2005-06-03 | ||
| GB0610513A GB0610513D0 (en) | 2006-05-26 | 2006-05-26 | Novel compounds |
| GB0610513.4 | 2006-05-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006129100A1 true WO2006129100A1 (en) | 2006-12-07 |
Family
ID=38002449
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2006/002015 WO2006129100A1 (en) | 2005-06-03 | 2006-06-02 | Novel compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2006129100A1 (en) |
Cited By (59)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007085833A2 (en) * | 2006-01-26 | 2007-08-02 | Astrazeneca Ab | Pyrimidine derivatives |
| WO2009010789A2 (en) * | 2007-07-16 | 2009-01-22 | Astrazeneca Ab | Pyrimidine derivatives 934 |
| WO2009158571A1 (en) * | 2008-06-27 | 2009-12-30 | Avila Therapeutics And Uses Thereof | Heteroaryl compounds and uses thereof |
| JP2010526150A (en) * | 2007-05-04 | 2010-07-29 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Compounds and compositions as c-kit and PDGFR kinase inhibitors |
| CN101857572A (en) * | 2010-06-08 | 2010-10-13 | 东南大学 | 4-aromatic amino pyrimidine derivative and application thereof |
| WO2010138578A1 (en) * | 2009-05-27 | 2010-12-02 | Abbott Laboratories | Pyrimidine inhibitors of kinase activity |
| WO2011079051A1 (en) | 2009-12-23 | 2011-06-30 | Takeda Pharmaceutical Company Limited | Fused heteroaromatic pyrrolidinones as syk inhibitors |
| EP2387572A2 (en) * | 2009-01-15 | 2011-11-23 | Rigel Pharmaceuticals, Inc. | Protein kinase c inhibitors and uses thereof |
| EP2489663A1 (en) | 2011-02-16 | 2012-08-22 | Almirall, S.A. | Compounds as syk kinase inhibitors |
| US8338439B2 (en) | 2008-06-27 | 2012-12-25 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| WO2012177714A1 (en) | 2011-06-22 | 2012-12-27 | Takeda Pharmaceutical Company Limited | Substituted 6-aza-isoindolin-1-one derivatives |
| US20130023532A1 (en) * | 2010-03-26 | 2013-01-24 | Casillas Linda N | Indazolyl-pyrimidines as kinase inhibitors |
| US8551984B2 (en) | 2009-12-17 | 2013-10-08 | Merck Sharp & Dohme Corp. | Aminopyrimidines as SYK inhibitors |
| US8563568B2 (en) | 2010-08-10 | 2013-10-22 | Celgene Avilomics Research, Inc. | Besylate salt of a BTK inhibitor |
| CN103619170A (en) * | 2011-05-04 | 2014-03-05 | 默沙东公司 | Amino-pyridine-containing spleen tyrosine kinase (SYK) inhibitors |
| US8735417B2 (en) | 2009-12-17 | 2014-05-27 | Merck Sharp & Dohme Corp. | Aminopyrimidines as Syk inhibitors |
| US8796255B2 (en) | 2010-11-10 | 2014-08-05 | Celgene Avilomics Research, Inc | Mutant-selective EGFR inhibitors and uses thereof |
| WO2015003858A1 (en) * | 2013-07-12 | 2015-01-15 | Basf Se | Process for manufacturing haloacetamides |
| US8975249B2 (en) | 2010-11-01 | 2015-03-10 | Celgene Avilomics Research, Inc. | Heterocyclic compounds and uses thereof |
| US8987456B2 (en) | 2011-10-05 | 2015-03-24 | Merck Sharp & Dohme Corp. | 3-pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9006444B2 (en) | 2011-10-05 | 2015-04-14 | Merck Sharp & Dohme Corp. | Phenyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9012462B2 (en) | 2008-05-21 | 2015-04-21 | Ariad Pharmaceuticals, Inc. | Phosphorous derivatives as kinase inhibitors |
| US9056839B2 (en) | 2012-03-15 | 2015-06-16 | Celgene Avilomics Research, Inc. | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| US9108927B2 (en) | 2012-03-15 | 2015-08-18 | Celgene Avilomics Research, Inc. | Salts of an epidermal growth factor receptor kinase inhibitor |
| US9120785B2 (en) | 2011-05-10 | 2015-09-01 | Merck Sharp & Dohme Corp. | Pyridyl aminopyridines as Syk inhibitors |
| US9126950B2 (en) | 2012-12-21 | 2015-09-08 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| US9145387B2 (en) | 2013-02-08 | 2015-09-29 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| US9145391B2 (en) | 2011-05-10 | 2015-09-29 | Merck Sharp & Dohme Corp. | Bipyridylaminopyridines as Syk inhibitors |
| US9216173B2 (en) | 2011-10-05 | 2015-12-22 | Merck Sharp & Dohme Corp. | 2-Pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9238629B2 (en) | 2010-11-01 | 2016-01-19 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| US9242984B2 (en) | 2012-06-20 | 2016-01-26 | Merck Sharp & Dohme Corp. | Pyrazolyl derivatives as Syk inhibitors |
| US9273077B2 (en) | 2008-05-21 | 2016-03-01 | Ariad Pharmaceuticals, Inc. | Phosphorus derivatives as kinase inhibitors |
| US9290490B2 (en) | 2011-05-10 | 2016-03-22 | Merck Sharp & Dohme Corp. | Aminopyrimidines as Syk inhibitors |
| US9353070B2 (en) | 2012-08-17 | 2016-05-31 | Basf Se | Carbamat-benzoxazinones |
| US9353066B2 (en) | 2012-08-20 | 2016-05-31 | Merck Sharp & Dohme Corp. | Substituted phenyl-Spleen Tyrosine Kinase (Syk) inhibitors |
| US9359312B2 (en) | 2011-12-23 | 2016-06-07 | Basf Se | Process for manufacturing benzoxazinones |
| US9364476B2 (en) | 2011-10-28 | 2016-06-14 | Celgene Avilomics Research, Inc. | Methods of treating a Bruton's Tyrosine Kinase disease or disorder |
| US9376418B2 (en) | 2012-06-22 | 2016-06-28 | Merck Sharp & Dohme Corp. | Substituted pyridine spleen tyrosine kinase (SYK) inhibitors |
| US9415049B2 (en) | 2013-12-20 | 2016-08-16 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| US9416111B2 (en) | 2012-06-22 | 2016-08-16 | Merck Sharp & Dohme Corp. | Substituted diazine and triazine spleen tyrosine kinease (Syk) inhibitors |
| US9487504B2 (en) | 2012-06-20 | 2016-11-08 | Merck Sharp & Dohme Corp. | Imidazolyl analogs as syk inhibitors |
| US9492471B2 (en) | 2013-08-27 | 2016-11-15 | Celgene Avilomics Research, Inc. | Methods of treating a disease or disorder associated with Bruton'S Tyrosine Kinase |
| US9499534B2 (en) | 2013-04-26 | 2016-11-22 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminopyrimidines as spleen tyrosine kinase inhibitors |
| US9586931B2 (en) | 2012-09-28 | 2017-03-07 | Merck Sharp & Dohme Corp. | Triazolyl derivatives as Syk inhibitors |
| US9598405B2 (en) | 2012-12-21 | 2017-03-21 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminopyridines as spleen tyrosine kinase inhibitors |
| US9611283B1 (en) | 2013-04-10 | 2017-04-04 | Ariad Pharmaceuticals, Inc. | Methods for inhibiting cell proliferation in ALK-driven cancers |
| US9624210B2 (en) | 2012-12-12 | 2017-04-18 | Merck Sharp & Dohme Corp. | Amino-pyrimidine-containing spleen tyrosine kinase (Syk) inhibitors |
| US9670196B2 (en) | 2013-12-20 | 2017-06-06 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as Spleen Tyrosine Kinase inhibitors |
| US9745295B2 (en) | 2013-04-26 | 2017-08-29 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9775839B2 (en) | 2014-03-13 | 2017-10-03 | Merck Sharp & Dohme Corp. | 2-pyrazine carboxamides as spleen tyrosine kinase inhibitors |
| US9783531B2 (en) | 2013-12-20 | 2017-10-10 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9822107B2 (en) | 2013-12-20 | 2017-11-21 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9834571B2 (en) | 2012-05-05 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| US9834518B2 (en) | 2011-05-04 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| US9908884B2 (en) | 2009-05-05 | 2018-03-06 | Dana-Farber Cancer Institute, Inc. | EGFR inhibitors and methods of treating disorders |
| US10005760B2 (en) | 2014-08-13 | 2018-06-26 | Celgene Car Llc | Forms and compositions of an ERK inhibitor |
| WO2019076817A1 (en) | 2017-10-19 | 2019-04-25 | Bayer Animal Health Gmbh | Use of fused heteroaromatic pyrrolidones for treatment and prevention of diseases in animals |
| US20220098181A1 (en) * | 2005-06-08 | 2022-03-31 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the jak pathway |
| US11351168B1 (en) | 2008-06-27 | 2022-06-07 | Celgene Car Llc | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001060816A1 (en) * | 2000-02-17 | 2001-08-23 | Amgen Inc. | Kinase inhibitors |
| WO2004074244A2 (en) * | 2003-02-20 | 2004-09-02 | Smithkline Beecham Corporation | Pyrimidine compounds |
| US20040242578A1 (en) * | 2000-12-21 | 2004-12-02 | Amogh Boloor | Pyrimidineamines as angiogenesis modulators |
| WO2005026158A1 (en) * | 2003-09-16 | 2005-03-24 | Novartis Ag | 2,4 di (hetero) -arylamino-pyrimidine derivatives as zap-70 and/or syk inhibitors |
-
2006
- 2006-06-02 WO PCT/GB2006/002015 patent/WO2006129100A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001060816A1 (en) * | 2000-02-17 | 2001-08-23 | Amgen Inc. | Kinase inhibitors |
| US20040242578A1 (en) * | 2000-12-21 | 2004-12-02 | Amogh Boloor | Pyrimidineamines as angiogenesis modulators |
| WO2004074244A2 (en) * | 2003-02-20 | 2004-09-02 | Smithkline Beecham Corporation | Pyrimidine compounds |
| WO2005026158A1 (en) * | 2003-09-16 | 2005-03-24 | Novartis Ag | 2,4 di (hetero) -arylamino-pyrimidine derivatives as zap-70 and/or syk inhibitors |
Cited By (124)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20220098181A1 (en) * | 2005-06-08 | 2022-03-31 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the jak pathway |
| US11827628B2 (en) * | 2005-06-08 | 2023-11-28 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| AU2007209126B2 (en) * | 2006-01-26 | 2012-01-19 | Astrazeneca Ab | Pyrimidine derivatives |
| WO2007085833A3 (en) * | 2006-01-26 | 2007-09-27 | Astrazeneca Ab | Pyrimidine derivatives |
| WO2007085833A2 (en) * | 2006-01-26 | 2007-08-02 | Astrazeneca Ab | Pyrimidine derivatives |
| JP2010526150A (en) * | 2007-05-04 | 2010-07-29 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Compounds and compositions as c-kit and PDGFR kinase inhibitors |
| WO2009010789A2 (en) * | 2007-07-16 | 2009-01-22 | Astrazeneca Ab | Pyrimidine derivatives 934 |
| WO2009010789A3 (en) * | 2007-07-16 | 2009-05-07 | Astrazeneca Ab | Pyrimidine derivatives 934 |
| US7718653B2 (en) | 2007-07-16 | 2010-05-18 | Astrazeneca Ab | Pyrimidine derivatives for inhibiting Eph receptors |
| CN101796046A (en) * | 2007-07-16 | 2010-08-04 | 阿斯利康(瑞典)有限公司 | Pyrimidine derivatives 934 |
| US9273077B2 (en) | 2008-05-21 | 2016-03-01 | Ariad Pharmaceuticals, Inc. | Phosphorus derivatives as kinase inhibitors |
| US9012462B2 (en) | 2008-05-21 | 2015-04-21 | Ariad Pharmaceuticals, Inc. | Phosphorous derivatives as kinase inhibitors |
| US10596172B2 (en) | 2008-06-27 | 2020-03-24 | Celgene Car Llc | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US8710222B2 (en) | 2008-06-27 | 2014-04-29 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| AU2009262068C1 (en) * | 2008-06-27 | 2015-07-02 | Celgene Car Llc | Heteroaryl compounds and uses thereof |
| US9212181B2 (en) | 2008-06-27 | 2015-12-15 | Celgene Avilomics Research, Inc. | Substituted 2,4-diaminopyrimidines as kinase inhibitors |
| WO2009158571A1 (en) * | 2008-06-27 | 2009-12-30 | Avila Therapeutics And Uses Thereof | Heteroaryl compounds and uses thereof |
| US9296737B2 (en) | 2008-06-27 | 2016-03-29 | Celgene Avilomics Research, Inc. | Substituted 2,4-diaminopyrimidines as kinase inhibitors |
| JP2017137352A (en) * | 2008-06-27 | 2017-08-10 | セルジーン アビロミクス リサーチ, インコーポレイテッド | Heteroaryl compounds and uses thereof |
| US8338439B2 (en) | 2008-06-27 | 2012-12-25 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US10010548B2 (en) | 2008-06-27 | 2018-07-03 | Celgene Car Llc | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| JP2019194235A (en) * | 2008-06-27 | 2019-11-07 | セルジーン シーエーアール エルエルシー | Heteroaryl compounds and uses thereof |
| US11351168B1 (en) | 2008-06-27 | 2022-06-07 | Celgene Car Llc | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US8450335B2 (en) | 2008-06-27 | 2013-05-28 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US9987276B2 (en) | 2008-06-27 | 2018-06-05 | Celgene Car Llc | Substituted 2,4-diaminopyrimidines as kinase inhibitors |
| JP2011526299A (en) * | 2008-06-27 | 2011-10-06 | アビラ セラピューティクス, インコーポレイテッド | Heteroaryl compounds and their use |
| US10828300B2 (en) | 2008-06-27 | 2020-11-10 | Celgene Car Llc | Substituted 2,4-diaminopyrimidines as kinase inhibitors |
| US8609679B2 (en) | 2008-06-27 | 2013-12-17 | Celgene Avilomics Research, Inc. | 2,4-diaminopyrimidines useful as kinase inhibitors |
| AU2009262068B2 (en) * | 2008-06-27 | 2014-12-11 | Celgene Car Llc | Heteroaryl compounds and uses thereof |
| US9409921B2 (en) | 2008-06-27 | 2016-08-09 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines as kinase inhibitors |
| EP2387572A4 (en) * | 2009-01-15 | 2012-07-25 | Rigel Pharmaceuticals Inc | Protein kinase c inhibitors and uses thereof |
| US9095593B2 (en) | 2009-01-15 | 2015-08-04 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| CN102292333A (en) * | 2009-01-15 | 2011-12-21 | 里格尔药品股份有限公司 | Protein kinase c inhibitors and uses thereof |
| EP2387572A2 (en) * | 2009-01-15 | 2011-11-23 | Rigel Pharmaceuticals, Inc. | Protein kinase c inhibitors and uses thereof |
| US9453028B2 (en) | 2009-01-15 | 2016-09-27 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| JP2012515206A (en) * | 2009-01-15 | 2012-07-05 | ライジェル ファーマシューティカルズ, インコーポレイテッド | Protein kinase C inhibitors and uses thereof |
| CN102292333B (en) * | 2009-01-15 | 2015-05-13 | 里格尔药品股份有限公司 | Protein kinase c inhibitors and uses thereof |
| US9908884B2 (en) | 2009-05-05 | 2018-03-06 | Dana-Farber Cancer Institute, Inc. | EGFR inhibitors and methods of treating disorders |
| CN102459236B (en) * | 2009-05-27 | 2014-10-29 | Abbvie公司 | Pyrimidine inhibitors of kinase activity |
| WO2010138578A1 (en) * | 2009-05-27 | 2010-12-02 | Abbott Laboratories | Pyrimidine inhibitors of kinase activity |
| US8486933B2 (en) | 2009-05-27 | 2013-07-16 | Abbvie Inc. | Pyrimidine inhibitors of kinase activity |
| CN102459236A (en) * | 2009-05-27 | 2012-05-16 | 雅培制药有限公司 | Pyrimidine inhibitors of kinase activity |
| US8551984B2 (en) | 2009-12-17 | 2013-10-08 | Merck Sharp & Dohme Corp. | Aminopyrimidines as SYK inhibitors |
| US8735417B2 (en) | 2009-12-17 | 2014-05-27 | Merck Sharp & Dohme Corp. | Aminopyrimidines as Syk inhibitors |
| US8759366B2 (en) | 2009-12-17 | 2014-06-24 | Merck Sharp & Dohme Corp. | Aminopyrimidines as SYK inhibitors |
| US9108970B2 (en) | 2009-12-23 | 2015-08-18 | Takeda Pharmaceutical Company Limited | Fused heteroaromatic pyrrolidinones |
| EP3489236A1 (en) | 2009-12-23 | 2019-05-29 | Takeda Pharmaceutical Company Limited | Process for the preparation of fused heteroaromatic pyrrolidinones |
| EP3825316A1 (en) | 2009-12-23 | 2021-05-26 | Takeda Pharmaceutical Company Limited | Fused heteroaromatic pyrrolidinones as syk inhibitors |
| EP2902392A1 (en) | 2009-12-23 | 2015-08-05 | Takeda Pharmaceutical Company Limited | Fused heteroaromatic pyrrolidinones as syk inhibitors |
| WO2011079051A1 (en) | 2009-12-23 | 2011-06-30 | Takeda Pharmaceutical Company Limited | Fused heteroaromatic pyrrolidinones as syk inhibitors |
| US9181255B2 (en) | 2009-12-23 | 2015-11-10 | Takeda Pharmaceutical Company Limited | Fused heteroaromatic pyrrolidinones as SYK inhibitors |
| US8440689B2 (en) | 2009-12-23 | 2013-05-14 | Takeda Pharmaceutical Company Limited | Fused heteroaromatic pyrrolidinones |
| US20130023532A1 (en) * | 2010-03-26 | 2013-01-24 | Casillas Linda N | Indazolyl-pyrimidines as kinase inhibitors |
| CN101857572A (en) * | 2010-06-08 | 2010-10-13 | 东南大学 | 4-aromatic amino pyrimidine derivative and application thereof |
| US8563568B2 (en) | 2010-08-10 | 2013-10-22 | Celgene Avilomics Research, Inc. | Besylate salt of a BTK inhibitor |
| US9604936B2 (en) | 2010-08-10 | 2017-03-28 | Celgene Car Llc | Besylate salt of a BTK inhibitor |
| US9765038B2 (en) | 2010-11-01 | 2017-09-19 | Celgene Car Llc | Heteroaryl compounds and uses thereof |
| US10434101B2 (en) | 2010-11-01 | 2019-10-08 | Celgene Car Llc | Heterocyclic compounds and uses thereof |
| US9238629B2 (en) | 2010-11-01 | 2016-01-19 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| US10081606B2 (en) | 2010-11-01 | 2018-09-25 | Celgene Car Llc | Heteroaryl compounds and uses thereof |
| US9375431B2 (en) | 2010-11-01 | 2016-06-28 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidine compounds useful as kinase inhibtors |
| US8975249B2 (en) | 2010-11-01 | 2015-03-10 | Celgene Avilomics Research, Inc. | Heterocyclic compounds and uses thereof |
| US11096942B2 (en) | 2010-11-01 | 2021-08-24 | Celgene Car Llc | Heterocyclic compounds and uses thereof |
| US9867824B2 (en) | 2010-11-01 | 2018-01-16 | Celgene Car Llc | Heterocyclic compounds and uses thereof |
| US9409887B2 (en) | 2010-11-10 | 2016-08-09 | Celgene Avilomics Research, Inc. | Mutant-selective EGFR inhibitors and uses thereof |
| US8796255B2 (en) | 2010-11-10 | 2014-08-05 | Celgene Avilomics Research, Inc | Mutant-selective EGFR inhibitors and uses thereof |
| US9868723B2 (en) | 2010-11-10 | 2018-01-16 | Celgene Car Llc | Mutant-selective EGFR inhibitors and uses thereof |
| EP2489663A1 (en) | 2011-02-16 | 2012-08-22 | Almirall, S.A. | Compounds as syk kinase inhibitors |
| US9834518B2 (en) | 2011-05-04 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| CN103619170B (en) * | 2011-05-04 | 2016-07-06 | 默沙东公司 | Spleen tyrosine kinase (SYK) inhibitor containing amino-pyridine |
| CN103619170A (en) * | 2011-05-04 | 2014-03-05 | 默沙东公司 | Amino-pyridine-containing spleen tyrosine kinase (SYK) inhibitors |
| US9145391B2 (en) | 2011-05-10 | 2015-09-29 | Merck Sharp & Dohme Corp. | Bipyridylaminopyridines as Syk inhibitors |
| US9290490B2 (en) | 2011-05-10 | 2016-03-22 | Merck Sharp & Dohme Corp. | Aminopyrimidines as Syk inhibitors |
| US9120785B2 (en) | 2011-05-10 | 2015-09-01 | Merck Sharp & Dohme Corp. | Pyridyl aminopyridines as Syk inhibitors |
| US9663514B2 (en) | 2011-06-22 | 2017-05-30 | Takeda Pharmaceutical Company Limited | Substituted 6-aza-isoindolin-1-one derivatives |
| WO2012177714A1 (en) | 2011-06-22 | 2012-12-27 | Takeda Pharmaceutical Company Limited | Substituted 6-aza-isoindolin-1-one derivatives |
| US9056873B2 (en) | 2011-06-22 | 2015-06-16 | Takeda Pharmaceutical Company Limited | Substituted 6-aza-isoindolin-1-one derivatives |
| US8987456B2 (en) | 2011-10-05 | 2015-03-24 | Merck Sharp & Dohme Corp. | 3-pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9006444B2 (en) | 2011-10-05 | 2015-04-14 | Merck Sharp & Dohme Corp. | Phenyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9216173B2 (en) | 2011-10-05 | 2015-12-22 | Merck Sharp & Dohme Corp. | 2-Pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9364476B2 (en) | 2011-10-28 | 2016-06-14 | Celgene Avilomics Research, Inc. | Methods of treating a Bruton's Tyrosine Kinase disease or disorder |
| US9359312B2 (en) | 2011-12-23 | 2016-06-07 | Basf Se | Process for manufacturing benzoxazinones |
| US10570099B2 (en) | 2012-03-15 | 2020-02-25 | Celgene Car Llc | Salts of an epidermal growth factor receptor kinase inhibitor |
| US9539255B2 (en) | 2012-03-15 | 2017-01-10 | Celgene Avilomics Research, Inc. | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| US9056839B2 (en) | 2012-03-15 | 2015-06-16 | Celgene Avilomics Research, Inc. | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| US9108927B2 (en) | 2012-03-15 | 2015-08-18 | Celgene Avilomics Research, Inc. | Salts of an epidermal growth factor receptor kinase inhibitor |
| US10005738B2 (en) | 2012-03-15 | 2018-06-26 | Celgene Car Llc | Salts of an epidermal growth factor receptor kinase inhibitor |
| US9540335B2 (en) | 2012-03-15 | 2017-01-10 | Celgene Avilomics Research, Inc. | Salts of an epidermal growth factor receptor kinase inhibitor |
| US10004741B2 (en) | 2012-03-15 | 2018-06-26 | Celgene Car Llc | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| US11292772B2 (en) | 2012-03-15 | 2022-04-05 | Celgene Car Llc | Salts of an epidermal growth factor receptor kinase inhibitor |
| US10946016B2 (en) | 2012-03-15 | 2021-03-16 | Celgene Car Llc | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| US9834571B2 (en) | 2012-05-05 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| US9242984B2 (en) | 2012-06-20 | 2016-01-26 | Merck Sharp & Dohme Corp. | Pyrazolyl derivatives as Syk inhibitors |
| US9487504B2 (en) | 2012-06-20 | 2016-11-08 | Merck Sharp & Dohme Corp. | Imidazolyl analogs as syk inhibitors |
| US9416111B2 (en) | 2012-06-22 | 2016-08-16 | Merck Sharp & Dohme Corp. | Substituted diazine and triazine spleen tyrosine kinease (Syk) inhibitors |
| US9376418B2 (en) | 2012-06-22 | 2016-06-28 | Merck Sharp & Dohme Corp. | Substituted pyridine spleen tyrosine kinase (SYK) inhibitors |
| US9353070B2 (en) | 2012-08-17 | 2016-05-31 | Basf Se | Carbamat-benzoxazinones |
| US9353066B2 (en) | 2012-08-20 | 2016-05-31 | Merck Sharp & Dohme Corp. | Substituted phenyl-Spleen Tyrosine Kinase (Syk) inhibitors |
| US9586931B2 (en) | 2012-09-28 | 2017-03-07 | Merck Sharp & Dohme Corp. | Triazolyl derivatives as Syk inhibitors |
| US9624210B2 (en) | 2012-12-12 | 2017-04-18 | Merck Sharp & Dohme Corp. | Amino-pyrimidine-containing spleen tyrosine kinase (Syk) inhibitors |
| US9549927B2 (en) | 2012-12-21 | 2017-01-24 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| US9126950B2 (en) | 2012-12-21 | 2015-09-08 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| US9598405B2 (en) | 2012-12-21 | 2017-03-21 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminopyridines as spleen tyrosine kinase inhibitors |
| US9145387B2 (en) | 2013-02-08 | 2015-09-29 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| US9561228B2 (en) | 2013-02-08 | 2017-02-07 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| US9980964B2 (en) | 2013-02-08 | 2018-05-29 | Celgene Car Llc | ERK inhibitors and uses thereof |
| US9504686B2 (en) | 2013-02-08 | 2016-11-29 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| US9796700B2 (en) | 2013-02-08 | 2017-10-24 | Celgene Car Llc | ERK inhibitors and uses thereof |
| US9611283B1 (en) | 2013-04-10 | 2017-04-04 | Ariad Pharmaceuticals, Inc. | Methods for inhibiting cell proliferation in ALK-driven cancers |
| US9499534B2 (en) | 2013-04-26 | 2016-11-22 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminopyrimidines as spleen tyrosine kinase inhibitors |
| US9745295B2 (en) | 2013-04-26 | 2017-08-29 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| CN105452214A (en) * | 2013-07-12 | 2016-03-30 | 巴斯夫欧洲公司 | Process for manufacturing haloacetamides |
| US9546130B2 (en) | 2013-07-12 | 2017-01-17 | Basf Se | Process for manufacturing haloacetamides |
| WO2015003858A1 (en) * | 2013-07-12 | 2015-01-15 | Basf Se | Process for manufacturing haloacetamides |
| US9492471B2 (en) | 2013-08-27 | 2016-11-15 | Celgene Avilomics Research, Inc. | Methods of treating a disease or disorder associated with Bruton'S Tyrosine Kinase |
| US9670196B2 (en) | 2013-12-20 | 2017-06-06 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as Spleen Tyrosine Kinase inhibitors |
| US9415049B2 (en) | 2013-12-20 | 2016-08-16 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| US9822107B2 (en) | 2013-12-20 | 2017-11-21 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9783531B2 (en) | 2013-12-20 | 2017-10-10 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9775839B2 (en) | 2014-03-13 | 2017-10-03 | Merck Sharp & Dohme Corp. | 2-pyrazine carboxamides as spleen tyrosine kinase inhibitors |
| US10202364B2 (en) | 2014-08-13 | 2019-02-12 | Celgene Car Llc | Forms and compositions of an ERK inhibitor |
| US10005760B2 (en) | 2014-08-13 | 2018-06-26 | Celgene Car Llc | Forms and compositions of an ERK inhibitor |
| WO2019076817A1 (en) | 2017-10-19 | 2019-04-25 | Bayer Animal Health Gmbh | Use of fused heteroaromatic pyrrolidones for treatment and prevention of diseases in animals |
| US11077111B2 (en) | 2017-10-19 | 2021-08-03 | Bayer Animal Health Gmbh | Use of fused heteroaromatic pyrrolidones for treatment and prevention of diseases in animals |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006129100A1 (en) | Novel compounds | |
| EP2376481B1 (en) | Pyrimidinecarboxamide derivatives as inhibitors of syk kinase | |
| JP6663857B2 (en) | Pyrazolopyridine and pyrazolopyrimidine | |
| WO2007028445A1 (en) | 6-indolyl-4-yl-amino-5-halogeno-2-pyrimidinyl-amino derivatives | |
| EP2142516B1 (en) | Pyrimidine hydrazide compounds as pgds inhibitors | |
| WO2007009681A1 (en) | 1 , 1-DIOXID0-2 , 3-DIHYDRO-l , 2-BENZISOTHIAZ0L-6-YL-1H-INDAZOL-4-YL-2 , 4-PYRIMIDINEDI AMINE DERIVATIVES | |
| TWI680967B (en) | New heteroaryls and their use as medicaments | |
| KR102228764B1 (en) | Heterocyclic amides as kinase inhibitors | |
| EP2613781B1 (en) | Indazole derivatives for use in the treatment of influenza virus infection | |
| ES2325035T3 (en) | ERBB QUINASE INHIBITORS OF 2-PYRIMIDINYL PYRAZOLOPIRIDINE. | |
| WO2007085540A1 (en) | 1h-indaz0l-4-yl-2 , 4-pyrimidinediamine derivatives | |
| ES2409057T3 (en) | Alkyl compounds substituted with heteroaryl and method of use | |
| KR101940831B1 (en) | (alpha-substituted aralkylamino and heteroarylalkylamino)pyrimidinyl and 1,3,5-triazinyl benzimidazoles, pharmaceutical compositions containing them, and these compounds for use in treating proliferative diseases | |
| US8785445B2 (en) | 7-phenoxychroman carboxylic acid derivatives | |
| TW200800215A (en) | Novel compounds | |
| EP2351745A1 (en) | Pyrazine derivatives and use as PI3K inhibitors | |
| CA2589793A1 (en) | 4-(4-(imidazol-4-yl)pyrimidin-2-ylamino) benzamides as cdk inhibitors | |
| JP2007522142A (en) | Benzimidazole-substituted thiophene derivatives having activity against IKK3 | |
| WO2014195400A1 (en) | Kinase inhibitors | |
| CA2640665A1 (en) | Pyridinone pyrazole urea and pyrimidinone pyrazole urea derivatives | |
| SK9162001A3 (en) | Substituted (aminoiminomethyl or aminomethyl)benzoheteroaryl compounds as factor xa inhibitors | |
| WO2016203335A1 (en) | Novel pyrido[2,3-b]pyrazinones as bet-family bromodomain inhibitors | |
| CN117858707A (en) | CBL-B modulators and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06744069 Country of ref document: EP Kind code of ref document: A1 |