PROCESS FOR PREPARING GATIFLOXACIN
Field of the technique This invention relates to a simplified process for preparing gatifloxacin, which avoids isolation of the intermediate compounds (denominated "one pot" process) .
Prior state of the art Gatifloxacin is the international common name of l-cyclopropyl-6-fluoro-l, 4-dihydro-8-methoxy- 1- (3-methyl-l-piperazinyl) -4-oxo-3-guinolin-carboxylic acid of formula (I) , with application in medicine and known for its antibiotic activity:
European patent application EP-A-230295 discloses a process for obtaining gatifloxacin that consists on the reaction of compound (II) with 2-
In this process the gatifloxacin is isolated in the form of a hemihydrate after a laborious process
of column chromatography and recrystallisation in methanol, which contributes towards making the final yield lower than 20% by weight. Moreover, in said process an undesired by-product is formed, resulting from demethylation at position 8 of the ring. European patent application EP-A-241206 discloses a process for preparing gatifloxacin, whose final steps are as follows:
Gatifloxacin (I)
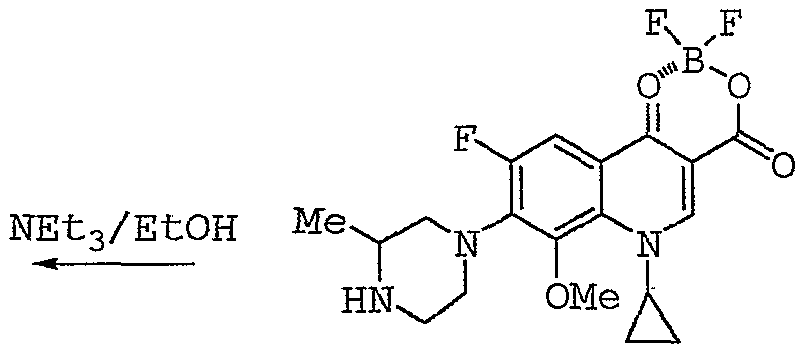
(IV) This process uses the intermediate compound (III) , which has been prepared and isolated in a separate operation, while the intermediate compound (IV) is also isolated before proceeding to its conversion into gatifloxacin by treatment with ethanol in the presence of triethylamine. The overall yield from these three steps is lower than 40%. These disadvantages — a synthesis involving several steps, low yields, and the need to isolate the intermediate products — hinder the production of gatifloxacin on an industrial scale.
There is therefore a need to provide a process for preparing gatifloxacin with a good chemical yield, without the need to isolate the intermediate compounds and that substantially avoids demethylation in position 8 of the ring. The processes termed in English "one pot" are characterised in that the synthesis is carried out in the same reaction vessel, without isolating the intermediate compounds, and by means of successive addition of the reacting compounds. The authors of the present invention have discovered a simplified process for preparing gatifloxacin which does not require isolation of the intermediate compounds .
Object of the invention The object of this invention is a simplified process for preparing gatifloxacin which does not require isolation of the intermediate compounds .
Detailed description of the invention The process object of the invention consists on reacting the compound (II)
with a silylating agent of formula ZSiR
3, in which Z is Me
3SiNH- or C1-, and R is either a C1-C4 alkyl chain or phenyl, within an aprotic polar solvent, for obtaining the intermediate compound (V) ,
in which R is either a Cχ-C
4 alkyl chain or phenyl, which without being isolated is then reacted with a boron trifluoride compound to prepare the intermediate compound (III) ,
which is not isolated and is then reacted with 2- ethylpiperazin after adjusting the pH to a value in the range of 8.5 to 9.5, to obtain the intermediate compound (IV) ,
which without being isolated is reacted with a C1-C chain alcohol in order to prepare gatifloxacin (I) . The compound (II) can be prepared according to the process described in preparation no. 6 of European patent application EP-A-241206. The silylating agents of formula ZSiR
3, in which Z is MeaSi H- o C1-, and R is either a C1.-C4 alkyl chain or phenyl, are compounds which can convert the hydroxyl group of a carboxylic acid into a
trialkyl, triarylsilyl or triarylalkylsilyl group in order to protect it or to activate it in subsequent reactions. The silylating agents can be chosen from the group of compounds such as: hexamethyldisilazane (HMDS) , and those described in the book "Protective Groups in Organic Synthesis" by T.W.Greene and P.G.M.Wuts, Third Edition, Editorial John Wiley & Sons, 1999 (ISBN 0-471-16019-9) (Pages 428-431) such as trimethylsilyl chloride, tert-butyldimethylsilyl chloride, isopropyldimethylsilyl chloride, di-tert- butyimethylsilyl chloride, phenyldimethylsilyl chloride, triisopropylsilyl chloride. Preferably, the silylating agent used is hexamethyldisilazane (HMDS) . The aprotic polar solvents are known for a skilled man in the art and are those organic solvents which have a certain polarity but which do not have acid hydrogen atoms . The book "Advanced Organic Chemistry" by J. March, 3rd Edition, published by John Wiley & Sons, 1985 (Table 13, page 319) , contains a list of solvents arranged by decreasing polarity, among which can be distinguished the following aprotic polar solvents: acetonitryl, dimethylsulphoxide di ethylformamide. Preferably, the aprotic polar solvent used is acetonitryl.
The boron trifluoride compound is a compound able to form chelates between oxygen atoms, and can be selected from the group of compounds such as: boron trifluoride, complex of boron trifluoride and diethyl ether (boron trifluoride ehtyletherate: BF3.Et20) , complex of boron trifluoride and tetrahydrofuran, tetrafluoroboric acid, complex of tetrafluoroboric acid and dimethyl ether. Preferably, the boron trifluoride compound used is the complex of boron trifluoride ethyletherate.
The C1-C4 alkyl alcohol can be selected from the group of compounds such as: methanol, ethanol, isopropanol, 1-propanol, 1-butanol, isobutanol, sec- butanol and tert-butanol. Preferably, the C1-C4 alcohol is methanol. The compound (II) reacts with the silylating agent at a temperature ranging between 70 and 85° C within the aprotic polar solvent for a period of time ranging between 1 and 2 hours. The intermediate compound (V) reacts with the boron trifluoride compound within the aprotic polar solvent at a temperature ranging between 15 and 25 °C until the compound (V) is observed to have disappeared completely. At this point the pH of the mixture is adjusted with a base to a value between 8.5 and 9.5. The bases are compounds which give water an alkaline pH and can be both inorganic and organic. The inorganic bases include the alkaline or alkaline-earth bicarbonates and carbonates. The organic bases include the tertiary amines. The tertiary amines include, for example: triethylamine, tributylamine, 4- methylmorpholine and 4-dimethylaminopryrridine. Preferably, triethylamine is used. The intermediate compound (III) reacts with
2-methylpiperazine within the aprotic polar solvent at a pH between 8.5 and 9.5, keeping the temperature within a range of 15 to 25° C for approximately 3 hours, until complete conversion of the compound (III) is observed. Distillation at low pressure is then carried out until a stirrable paste is obtained. The C1-C4 alkyl alcohol is added to that paste in order to remove the boron chelate, at a temperature ranging between 60 and 70° C, for the time
necessary to achieve the completion of the reaction of compound ( IV) , approximately 5 hours . Once the boron chelate elimination reaction has been completed the mixture is cooled, first to a temperature ranging between 25 and 35° C, and then to between 0 and 5° C. The precipitate obtained is gatifloxacin (I) , which is filtered, washed with the cold C1-C4 alcohol and dried at 40° C until a constant weight is obtained. Eventually, the crude gatifloxacin obtained can be recrystallised in methanol. Surprisingly, it has been found that the new process allows gatifloxacin to be obtained with a yield exceeding 75% on the basis of the compound (II) , without being needed to isolate the intermediates involved in the various synthetic steps, with a purity and a quality suitable for preparing pharmaceutical formulations, and with a content of less than 0.1% of the by-product resulting from demethylation in position 8 of the ring. The example which follows below is set out solely for the purposes of providing a detailed explanation of the process object of the invention for the skilled men in the art.
Example 1: Preparing gatifloxacin from compound (II) 10 g (0.0339 moles, 1 equivalent) of compound
(II) is placed in a flask, 30 ml of acetonitryl (3 volumes) is added and this is heated to a temperature of 76-80° C. Once reflux has been attained, and being the temperature maintained, 3.28 g (0.0203 moles, 0.6 equivalents) of hexamethyldisilazane (HMDS) is added with a compensated adding funnel. Once addition is
completed, the reaction is maintained with stirring for 1 hour at a temperature of 76-80° C. Once this period has elapsed, the reaction mixture is cooled to a temperature ranging between 0 and 15° C, and 5.78 g (0.0407 moles, 1.2 equivalents) of boron trifluoride ethyletherate is added while keeping the temperature below 15° C. Once addition is completed, the temperature is allowed to rise to 15- 25° C and it is kept under these conditions for approximately 2 hours. The pH of the mixture is then adjusted to an approximate value of 9 with triethylamine (approximately 2 ml) . To the resulting suspension is added a solution of 10.19 g (0.1017 moles, 3 equivalents) of 2-methylpiperazine in 28 ml of acetonitryl, while maintaining the temperature between 15 and 25° C. The resulting amber solution is kept with stirring under these conditions for approximately 3 hours . Once the reaction has been completed, the solution is distilled at low pressure until a stirrable paste is obtained. At this point 50 ml of methanol is added, the resulting suspension is raised to a temperature of 63-67° C and is kept under these conditions for approximately 5 hours . Once the reaction has been completed, the mixture is cooled to a temperature of 25-35° C in a water bath, and then at a temperature of 0-5° C in a water/ice bath for a further 1 hour. The resulting precipitate is filtered, washed with cold methanol (2 x 10 ml) and dried at 40° C in a vacuum oven to constant weight. 10.70 g of crude gatifloxacin is obtained, having a water content of 2.95% by weight. The yield of the process is 81.8%.
The crude product is crystallised in methanol by dissolving 20 g of crude gatifloxacin in 1 1 of
methanol (50 volumes) at a temperature of 63-67° C. Once all the product has been dissolved, the solution is left to cool to a temperature of 30-40° C, and then to a temperature of 0-5° C in a water/ice bath, maintaining it under these conditions for 1 hour. The resulting suspension is filtered and the solid retained is washed with 20 ml (1 volume) of cold methanol. The solid obtained is dried at 40° C in a vacuum oven to provide 18.65 g of gatifloxacin with a water content of 2.36% by weight.
The overall yield from the compound (II) is 77.7%, with a purity exceeding 99.8% as determined by HPLC chromatography. The content of by-product resulting from demethylation in position 8 of the ring is lower than 0.1% as determined by HPLC chromatography.










