WO2002009758A2 - Inhibitors of cellular efflux pumps of microbes - Google Patents
Inhibitors of cellular efflux pumps of microbes Download PDFInfo
- Publication number
- WO2002009758A2 WO2002009758A2 PCT/IN2001/000139 IN0100139W WO0209758A2 WO 2002009758 A2 WO2002009758 A2 WO 2002009758A2 IN 0100139 W IN0100139 W IN 0100139W WO 0209758 A2 WO0209758 A2 WO 0209758A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oxo
- dihydro
- fluoro
- salts
- amino
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/542—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame ortho- or peri-condensed with heterocyclic ring systems
- A61K31/545—Compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins, cefaclor, or cephalexine
- A61K31/546—Compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins, cefaclor, or cephalexine containing further heterocyclic rings, e.g. cephalothin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/65—Tetracyclines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
- C07D215/22—Oxygen atoms attached in position 2 or 4
- C07D215/233—Oxygen atoms attached in position 2 or 4 only one oxygen atom which is attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D455/00—Heterocyclic compounds containing quinolizine ring systems, e.g. emetine alkaloids, protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine
- C07D455/03—Heterocyclic compounds containing quinolizine ring systems, e.g. emetine alkaloids, protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine containing quinolizine ring systems directly condensed with at least one six-membered carbocyclic ring, e.g. protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine
- C07D455/04—Heterocyclic compounds containing quinolizine ring systems, e.g. emetine alkaloids, protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine containing quinolizine ring systems directly condensed with at least one six-membered carbocyclic ring, e.g. protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine containing a quinolizine ring system condensed with only one six-membered carbocyclic ring, e.g. julolidine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- This invention relates to compounds with efflux pump inhibitor properties, and which are therefore compounds which inhibit cellular efflux pumps of bacteria or other microbes.
- efflux pump inhibitors are useful, for example, against antibiotic-resistant microbial pathogens, for treating microbial infections by reducing the export of a co-administered antimicrobial agent or by preventing the export of a compound synthesized by microbes (e.g. bacteria, fungi) to allow or improve their growth.
- this invention also provides processes for preparation of such efflux pump inhibitors, compositions which include such efflux pump inhibitors, and the use of the compounds and compositions in methods for treatment of microbial infections.
- Microbes are known to have developed the ability to evolve different mechanisms of self-defense against antimicrobial agents.
- bacterial and fungal pathogens have developed mechanisms of resistance to antibiotics and antimicrobial agents used to inhibit their growth, or to treat infections by them in humans, animals and tissue cultures.
- treatment regimens can be adversely affected or, in some cases rendered ineffective.
- Multidrug efflux as a major cause of intrinsic drug resistance in many microorganisms, or overproduction of intrinsic pumps, or acquisition of pump genes from external sources, all play a prominent role, often resulting in high levels of resistance.
- MexAB-OprM, MexCD-OprJ, MexEF-OprN, MexXY-OprM, AcrAB-TolC, AcrEF, MarA, SoxS, or/and Tet pump/s are known to be present in Gram negative organisms such as P. aeruginosa and E. coli and are reviewed in recent publications and papers, such as K. K. Y. Wong et al, J. of Bacteriology, 2001, 183:367-374; K.
- Efflux transporters are also among different mechanisms responsible for the resistance to antibiotics displayed by gram-positive bacteria and mycobacteria, particularly aerobic gram- positive cocci.
- the multidrug transporter NorA belonging to the major facilitator superfamily (MFS) transporters, contributes to the resistance of Staphylococcus aureus to fluoroquinolone antibiotics.
- MFS major facilitator superfamily
- a minireview describes effllux-mediated resistance to fluoroquinolones in Gram- positive bacteria and the mycobacteria (K. Poole, Antimicro. Agents and Chemother., 2000, 44: 2595-2599).
- MFS transporters also belong the Bmr and QacA/QacB efflux pumps of Gram-positive bacteria, and EmrB of E. coli (H.
- MFP membrane-fusion protein
- Mef efflux system mediates large fractions of erythromycin-resistant clinical isolates of S. pneumoniae (N. J. Johnston et al, Antimicrob. Agents and Chemother., 1998, 42:2425; T. Nishijima et al, J. Antimicrob. Chemother., 1999, 43:637).
- Beta-haemolytic streptococci and pneumococci resistant to erythromycin due to the prsence of Mef A efflux pumps in Streptococcus pyogenes, Mef E pumps in S. pneumoniae, and an M phenotype bearing S. agalactiae possessing Mef A or Mef E pumps are found to be emergent and prevalent in Europe (C. Arpin et al, J. Antimicrob. Chemother., 1999, 44:133-138; E. Giovanetti et al, Antimicrob. Agents and Chemother., 1999, 43:1935-1940).
- One strategy to target resistance mechanisms of microbial self-defense is to find inhibitors of microbial efflux pumps and, in particular of bacterial and fungal efflux pumps.
- Patent WO 99/17791 discloses a method for inhibiting the selection or propagation of a bacterial mutant that overexpresses an efflux pump wherein the inhibitor disclosed is the dipeptide amide, L-phenylalanyl-L-arginyl-beta-naphthylamide (MC- 207,110), which is unlike the efflux pump inhibitor compounds of the present invention.
- US Patent 6,245,746, US Patent 6,114,310 and WO 9937667 all with US priority in US application 09/012,363 discloses methods of using efflux pump inhibitors which increase the susceptibility of microbes, in particular P. aeruginosa strains, to antimicrobial agents and pharmaceutical compositions including such compounds which are unlike the efflux inhibitor compounds of the present invention.
- US Patent 6,204,729 describes peptidomimetic, secondary amide containing benzoxazole derivatives as efflux pump inhibitors, methods of using such efflux pump inhibitor compounds and pharmaceutical compositions including such compounds which are unlike the efflux inhibitor compounds of the present invention.
- Patent WO 00/32196 discloses inhibitors of multidrug transport proteins which may be used in combination with existing antibacterial agents and/or antifungal agents, wherein the inhibitor is an indole or a urea or an aromatic amide or a quinoline, all of which inhibitors are unlike the efflux pump inhibitor compounds of the present invention.
- the inhibitors disclosed in patent WO 00/32196 are specifically inhibitors of bacteria expressing a norA pump, or a fungus expressing a multidrug transport protein.
- Novel inhibitors of the NorA multidrug transporter of S. aureus having structurally diverse chemical structures were also described by P. N. Markham et al, (Antimicrob. Agents and Chemother., 1999, 43:2404), among which the more active compounds include (a) those containing an indole moiety like the previously known inhibitor, reserpine, (b) biphenyl urea derivatives, (c) a substituted pyrimidinone derivative and (d) compounds INF 240 and INF 277, but they are all unlike the efflux pump inhibitor compounds of the present invention.
- Another inhibitor of the NorA MDR pump in a pathogenic S. aureus strain is 5'- methoxyhydnocarpin (F. R. Stermitz et al., Proc. New York Acad. of Sci., 2000, 97:1433), which has a structure unlike the efflux pump inhibitor compounds of the present invention.
- Nocardamin a cyclopeptide, was found to be a general antagonist of a tetracycline efflux pump from S. aureus. It has a structure unlike the efflux pump inhibitor compounds of the present invention.
- Minocycline and l,l-dimethyl-5-(l-hydroxypropyl)-4,6,7-trimethylindan (Ro 07-3149) inhibit the active tetracycline efflux pump in S. aureus 743 (T.Hirata et al., Biol Pharm Bull, 1998, 21:678). Both the compounds have a structure unlike the efflux pump inhibitor compounds of the present invention.
- Reserpine is not a usable compound for therapy because of its neurotoxicity at the concentration required for efflux pump inhibition.
- the inhibitors of single drug and multidrug transporters such as the dipeptide amide, MC- 207,110, are broad in specificity, inhibiting all three RND systems of P. aeruginosa involved in fluoroquinolone efflux, but have not been shown to be effective against pumps of other strains, for instance a NorA pump.
- the methods employed to demonstrate their efflux pump inhibitory properties are mainly in vitro methods. For demonstration of in vivo activity, recourse has had to be taken to parenteral administration ( cf. T.E.Renau et al, J.Med.Chem.,1999, 42, 4928), rather than oral administration, as it is generally known that oral bioavailability is poor for compounds of a peptidic nature.
- Potent inhibitors of microbial efflux pumps is thus an important goal for the improved control of infectious diseases, allowing a renaissance for drugs that are no longer effective owing to their efflux (K. Poole, Journal of Pharmacy and Pharmacology, 2001, 53:283-294).
- the current inventors have synthesised, screened and identified novel inhibitors of cellular efflux pumps of microbes. Distinctive structural features characterise the different sets of efflux pump inhibitors for different microorganisms as will be described in the following description.
- the present invention describes compounds of the formula I as novel microbial efflux pump inhibitors
- Rs H, Ci.s alkyl, amino, alkylamino, or acylamino.
- Re H, C ⁇ -6 alkyl, halo such as F, Cl, Br, or I, amino, or hydroxy;
- R 7 OH, halo such as F, Cl, Br, or I, or
- NR 9 R 10 wherein R 9 and Rj 0 are the same or different and represent H, . 6 alkyl or (CH 2 ) n OA, or R 9 is H and R ⁇ 0 is a 4-membered, 5-membered, 6-membered, or 7-membered carbocyclic, mono or bicyclic ring, or mono or bicyclic heterocyclic ring linked to the nitrogen of NR 9 R 10 through an atom of the heterocycle other than the heterocyclic atom, or R 9 and R ⁇ 0 taken together with the nitrogen atom to which they are attached form part of a heterocycle which heterocycle is monocyclic or bicyclic.
- the carbocycle and the heterocycle are optionally substituted at any position of the carbocyclic or heterocyclic group with COOR 3 CONHR 13 , OA, C ⁇ _ 6 alkyl, C 3 -C 6 cycloalkyl, aralkyl, trifluoroalkyl, substituted C ⁇ alkyl, or N R ⁇ R ⁇ 7 .
- Substituents of the alkyl group are selected from OA, NR 16 R ⁇ , or a halogen atom.
- R 16 and R ⁇ are the same or different and represent H or C .
- R 16 and R 16 and R J7 is C 3 -C 6 cycloalkyl, or substituted C ⁇ _ 6 alkyl, or R 16 and R ⁇ 7 taken together with the nitrogen atom form a heterocycle.
- R 16 or R i7 is a substituted alkyl group, the substituent is selected from NRi ⁇ R ⁇ , alkanoyl or aminoalkanoyl,
- R 7 NHOA, NHCOORrange, or NH(CH 2 ) radicalNR 9 Ri 0 ;
- this R 7 moiety is linked either to 2 core molecules of the Formula I to form a bis compound or the R 7 moiety has one of its link bonds linked to the core formula of Formula I and the second of its link bonds is linked to a phenyl carboxylic acid or ester moiety thereof, the phenyl carboxylic acid or ester being optionally substituted by the usual aromatic substituents, such as C C 3 alkyl linear or branched, aralkyl such as benzyl, amino, alkylamino, alkanoylamino, oc-aminoalkanoylamino, hydroxy, alkoxy, alkanoyloxy, oc-aminoalkanoyloxy, or halogen atoms, such as fluoro, chloro, bromo.
- A H, C 1 -6 alkyl, glycosyl, aralkyl, C ⁇ . 6 alkanoyl or aminoalkanoyl.
- the aminoalkanoyl group may be an aminoacid residue derived from one of the one of the 20 naturally occurring amino acids or the optically active isomers thereof, or the racemic mixtures thereof.
- the amino residue is derived from alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine or valine.
- A may also be C 6 H u O 6 , SO 3 H, or PO 3 H 2 .
- X CH, C-F, C-Cl, C-CH 3 , C-CF 3 , C-OCH 3 , C-OCHF 2 , C-OCF 3 , N or when X is equal to C it forms together with the nitrogen atom of the adjacent ring an optionally substituted 4-membered ring, 5-membered ring, 6-membered ring, 7-membered ring, containing carbon atoms and optionally one or more Y atoms representing one or more nitrogen, oxygen or sulfur atoms, such ring being further optionally substituted by a C ⁇ _ 6 alkyl group;
- the absolute stereochemistry can be either R- or S- configuration and any combination of configurations. Even racemic materials and diastereomers fulfil the structural generic descriptions.
- Another object of the invention is to provide a process for preparing the novel efflux pump inhibitors of the formula I of the invention.
- a further object of the invention is to provide pharmaceutical compositions comprising the compounds of the invention.
- a composition contains one or more compounds of formula I or salt, hydrate, polymorph or pseudopolymorph therefore.
- a composition of this invention may also include another antibiotic or antimicrobial compound.
- Yet another object of the invention relates to method of treatment of infections using the said compounds of the invention or compositions comprising them.
- Treatment comprises oral, parenteral administration and/or topical application of an effective amount of the compound of the invention or its compositions, whether single or in combination with an antibiotic or antimicrobial agent or two or more compounds of this invention.
- Yet a further object of the invention includes a method of suppressing growth of a bacterium or fungus expressing an efflux pump, comprising contacting said bacterium or fungus with an efflux pump inhibitor in the presence of a concentration of antibacterial or antifungal agent below the minimum inhibitory concentration (MIC) of said bacterium or fungus.
- Yet a further object of the invention includes methods for treating the infections in humans and animals, caused by sensitive and resistant microbial strains using an antimicrobial agent and an efflux pump inhibitor in an amount sufficient to reduce efflux pump activity, wherein said efflux pump inhibitor increases the susceptibility of said microbe to said antimicrobial agent.
- Yet a further object of the invention includes a method for prophylactic treatment of a human or animal, comprising administering to said human or animal at risk of a microbial infection an efflux pump inhibitor, wherein said efflux pump inhibitor decreases the pathogenicity of a microbe in said human or animal.
- Yet a further object of the invention includes a method for prophylactic treatment of human or animal, comprising administering to said human or animal at risk of a microbial infection an antimicrobial agent and an efflux pump inhibitor, wherein said efflux pump inhibitor increases the susceptibility of a microbe to said antimicrobial agent.
- Yet a further object of the invention includes a method of treatment using the efflux pump inhibitor compounds of the invention by administering, systemically or topically, optically pure compounds of the invention or stereochemically pure forms of the invention or their salts, hydrates, polymorphs, or pseudopolymorphs thereof to the affected human or animal, thereby avoiding the toxic effects associated with racemic mixtures of the compounds of the invention.
- Yet a further object of the invention is to enhance the antimicrobial activity of an antimicrobial agent against a microbe by contacting the microbe with an antimicrobial agent and an efflux pump inhibitor.
- the present invention describes heterocyclic compounds of the formula I as novel microbial efflux pump inhibitors
- Rs H, C ⁇ _s alkyl, amino, alkylamino, or acylamino.
- Re H, C ⁇ _ 6 alkyl, halo such as F, Cl, Br, or I, amino, or hydroxy;
- R 7 OH, halo such as F, Cl, Br, or I, or
- NR 9 R 10 wherein R 9 and Rio are the same or different and represent H, . 6 alkyl or (CH 2 ) n OA, or R 9 is H and Rio is a 4-membered, 5-membered, 6-membered, or 7-membered carbocyclic, mono or bicyclic ring, or mono or bicyclic heterocyclic ring linked to the nitrogen of NR 9 R ⁇ 0 through an atom of the heterocycle other than the heterocyclic atom, or R 9 and R w taken together with the nitrogen atom to which they are attached form part of a heterocycle which heterocycle is monocyclic, or bicyclic.
- the ring is a ring such as 2- azetidinyl
- R ⁇ 0 is a 5-membered ring
- the ring is a ring such as 2- (or 3-) pyrrolidinyl, or 2- (or 3-) furyl, or 2-(or 3-) thienyl, or 2-(or 4-)imidazolyl, or oxazolyl or pyrazolyl, or thiazolyl
- R 10 is a 6-membered ring
- the ring is a ring such as 2-(or 3-, or 4-) piperidinyl, or 2-(or 3-) piperazinyl, or 2-(or 3-) morpholinyl or 2-(or 4-)pyrimidinyI
- R 10 is a 7-membered heterocycle it is preferred that the heterocycle is a heterocycle such as azepinyl, ox
- heterocycle is such as azetidine, pyrrolidine, furan, thiophene, imidazole, oxazole, pyrazole, thiazole, piperidine, piperazine, pyrimidine, azepine, oxazine, thiazine, or bicyclic such as isoquinoline, quinuclidine, amino-3-azabicyclo [3.1.0]hexane.
- the carbocycle and the heterocycle are optionally substituted at any position of the heterocyclic group with COOR 3 CONHR 13 , OA, C ⁇ . 6 alkyl, aralkyl, trifluoroalkyl, substituted alkyl, or N
- Substituents of the alkyl group are selected from OA, NR ⁇ 6 R ⁇ , or a halogen atom.
- R ⁇ 6 and R ⁇ are the same or different and represent H or C ⁇ . 6 alkyl, or where one of R i6 and R ⁇ is hydrogen and the other of Rj 6 and R J7 is C 3 -C 6 cycloalkyl, or substituted alkyl, or R ⁇ 6 and R ⁇ taken together with the nitrogen atom form a heterocycle.
- R 16 or R J7 is a substituted alkyl group
- n 0, 1, or 2
- this R 7 moiety is linked either to 2 core molecules of the Formula I to form a bis compound or the R 7 moiety has one of its link bonds linked to the core formula of Formula I and the second of its link bonds is linked to a phenyl carboxylic acid or ester moiety thereof, optionally substituted by the usual aromatic substituents, such as C 1 -C 3 alkyl linear or branched, aralkyl such as benzyl, amino, alkylamino, alkanoylamino, oc-aminoalkanoylamino, hydroxy, alkoxy, alkanoyloxy, cc-aminoalkanoyloxy, or halogen atoms, such as fluoro, chloro, bromo.
- A H, C 1 - 5 alkyl, glycosyl, aralkyl, C ⁇ . 6 alkanoyl or aminoalkanoyl.
- the aminoalkanoyl group may be an aminoacid residue derived from one of the one of the 20 naturally occurring amino acids or the optically active isomers thereof, or the racemic mixtures thereof.
- the amino residue is derived from alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, histidine, isoleucine, leucine, Iysine, methionine, phenylalanine, proline, serine threonine, trytophan, tyrosine or valine.
- A may also be C 6 HnO 6 , SO 3 H, or PO 3 H 2 ,
- R 11 H, C ⁇ exclusively6 alkyl, C3.6 cycloalkyl, or heterocyclic group such as a 4-membered ring, it is preferred that the ring is a ring such 2-azetidinyl, a 5-membered ring such as 2- (or 3-) pyrrolidinyl, or 2- (or 3-) furyl, or 2-(or 3-) thienyl, or 2-(or 4-) imidazolyl, or oxazolyl or pyrazolyl, or thiazolyl, or a 6- membered ring such as 2-(or 3-,or 4-) piperidinyl, or 2-(or 3-) piperazinyl, or 2-(or 3-) morpholinyl, or 2-(or 4-)pyrimidinyl, or a 7-membered heterocycle such as azepinyl, or oxazinyl, or thiazinyl, or a bicyclic heterocycle such as tetrahydroisoquinolin
- X CH, C-F, C-Cl, C-CH 3 , C-CF 3 , C-OCH 3 , C-OCHF 2 , C-OCF 3 , N or when X is equal to C it forms together with the nitrogen atom of the adjacent ring a 5-membered ring, 6-membered ring, 7-membered ring, optionally containing besides carbon atoms additional Y atoms representing one or more nitrogen , oxygen or sulfur atoms, such ring being further optionally substituted by a
- Ci.6 alkyl group and their pharmaceutically acceptable salts, hydrates, polymorphs and pseudopolymorphs.
- R, and R 4 . are combined together to form an oxo group and Re is hydrogen or fluorine.
- the absolute stereochemistry can be either R- or S- configuration and any combination of configuration. Even racemic materials and diastereomers fulfil the structural generic descriptions.
- alkyl refers to a branched or unbranched C C 6 aliphatic hydrocarbon group.
- aralkyl refers to a C C 6 alkyl group substituted with an aryl group which aryl group is defined below.
- aryl group is defined below.
- One example of an aralkyl group is a benzyl group.
- arylaminoalkyl refers to an aryl group as defined below that is bonded respectively through an NH, oxygen , or S(O) t to an alkyl group as defined above.
- aryl refers to an aromatic group which has at least one ring having conjugated ⁇ electron system and includes both carbocyclic aryl (e.g., phenyl, naphthyl) and heterocyclic aryl groups (e.g. pyridyl, pyrimidyl, pyrazinyl, thienyl, furyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl and oxazolyl).
- the aryl group is preferably 5 to 14 carbons, more preferably 5 to 10 carbons.
- Aryl moieties includes monocyclic, bicyclic, and tricyclic rings, where each ring has preferably five or six members and are 6 ⁇ (or 6 pi)-annelated ring system or substituted 6 ⁇ annelated ring systems composed of a mix of carbocyclic and heterocyclic units (e.g. benzo, pyrido, pyrimido, pyrazino, thieno, furano, pyrrolo, pyrazolo, imidazolo, thiazolo and oxazolo).
- 6 ⁇ - annelated ring system refers to a ring which has 6 ⁇ electron and is considered aromatic.
- the ring may have 6 items in its backbone, such as pyridine, pyrimidine, benzene, or it may have less than 6 items in its backbone, such as pyrrole, pyrazole, furan, thiazole, oxazole or thiophene.
- the aryl moiety may be optionally monosubstituted or disubstituted independently with lower linear or branched C ⁇ -C 6 alkyl or hydroxyl, alkoxy, alkylthio, halogen, haloalkyl, mercapto, amino, mono linear or branched C ⁇ -C 3 alkylamino, di linear or branched C ⁇ -C 3 alkylamino, phenyl, substituted phenyl or optionally substituted phenylamino wherein the phenyl is substituted by usual aromatic substituents, such as C ⁇ -C 3 alkyl linear or branched, aralkyl such as benzyl, amino, alkylamino, alkanoylamino, oc-aminoalkanoylamino, hydroxy, alkoxy, alkanoyloxy, oc- aminoalkanoyloxy, or halogen atoms, such as fluoro, chloro, or bromo.
- Alkylamino is for example NHCH 3 , N(CH 3 ) 2 , NHC 2 H S , N(C 2 H S ) 2 , NHC 3 H 7 , N(C 3 H 7 ) 2 and
- Alkanoyl is for example acetyl, propionyl, ethoxycarbonyl, t-butoxycarbonyl,
- -Oq is for example pyrrolidinylethyl, piperidinylethyl, morpholinylethyl
- the preferred acid addition salts are those of hydrochloride, hydrobromide, hydroiodide, sulphate, sulfonate, sulfamate, phosphate and salts of organic acids such as acetate, lactate, succinate, oxalate, maleate, fumarate, malate, tartrate, citrate, ascorbate, gluconate, benzoate, cinnamate, methane sulphonate and p-toluene sulphonate.
- Preferred alkali addition salts are lithium, sodium, potassium salts, and alkaline earth salts are magnesium, calcium salts, or ammonium, or organic amines such as diethanolamine, N-methylglucamine, guanidine or heterocyclic amines such as choline, piperidine, N-methyl-4-hydroxypiperidine, hydroxyethylpyrrolidine, hydroxyethylpiperidine, morpholine, hydroxyethylmorpholine, piperazine, N-methyl piperazine and the like or basic amino acids such as optically pure or racemic isomers of arginine, lysine, histidine, tryptophan and the like.
- organic amines such as diethanolamine, N-methylglucamine, guanidine or heterocyclic amines such as choline, piperidine, N-methyl-4-hydroxypiperidine, hydroxyethylpyrrolidine, hydroxyethylpiperidine, morpholine, hydroxyethylmorpho
- RI C 2 H S , C 3 H 5 , CH(CH 3 )CH 2 CH 2 SC 6 H 5 , C 6 H,(2-CF3), where (2-CF3) means that the CF3 group is attached at the 2 position,
- (4-F) means that the F atom is attached at the 4 position
- (2,4-F2) means that there are 2 fluorine atoms, one at the 2 position and the other at the 4 position.
- X..Y..N C-CH 2 CH 2 CH(CH 3 ), C-OCH 2 CH(CH 3 ), C-OCH 2 CH 2 CH(CH 3 )
- R 2 COOH, COOC 2 H 5 , COOCH(CH 3 ) 2 , COO(CH 2 ) 3 CH 3 , COOCH 2 C 6 H 5 , COOCH 2 COOC 2 H 5 , COO(CH 2 ) 2 -morpholino,
- COphe-lys-Ome where phe-Iys-Ome is the methylester of the dipeptide from phenylalanine and lysine.
- R 5 H, CH 3 , or NH 2;
- R 7 F, Br, pyrrolidin-3-yl-amino, pyrrolidin-3-alkoxycarbonylamino, piperidin-4-yl-amino, pentaalkylpiperidin-4-yl-alkylamino, [l°c,5cc,6oc]-3-azabicyclo[3.1.0]hex-6-yl-amino, quinucIidinyI-3-yl-amino, 3-aminopyrrolidinyl, 5-aminopyrrolidinyl, aminopyrroUdinyl optionally further mono/poly substituted with C C 6 alkyl, (mono/poly aminoalkanoyl)aminopyrrolidinyl; alkoxycarbonyl (mono/poly aminoalkanoyl) aminopyrroUdinyl; acetamidopyrrolidinyl optionally further mono/poly substituted with C ⁇ -C 6 alkyl, hydroxypyrroli
- X C-H, C-OCH 3 , C-F, N and when X is linked to N of the adjacent ring, it has the meanings as defined above for X..Y...N
- Suitable acids are hydrochloric, hydrobromic, hydroiodic, sulphuric, sulfamic, sulfonic, phosphoric, acetic, lactic, succinic, oxalic, maleic, fumaric, malic, tartaric, citric, ascorbic, gluconic, benzoic, cinnamic, methanesulfonic and p-toluenesulfonic acid.
- the pharmaceuticaUy acceptable cationic salts of compounds I may be prepared by conventional methods from the corresponding acids e.g. by reaction with about one equimolar amount of a base.
- suitable cationic salts are those of alkaU metals such as Uthium, sodium or potassium, alkaline earth metals such as magnesium or calcium or ammonium or organic amines such diethanolamine, N-methylglucamine, guanidine or heterocycUc amines such as choline, piperidine, N-methyl-4-hydroxypiperidine, hydroxyethylpyrrolidine, hydroxyethylpiperidine, morpholine, hydroxyethylmorphoUne, piperazine, N-methyl piperazine and the like or basic amino acids such as opticaUy pure or racemic isomers of arginine, lysine, histidine, tryptophan and the like.
- hydrates, pseudopolymorphs and polymorphs are prepared by methods known in the art. DetaUed descriptions of different methods to generate hydrates, solvates, pseudopolymorphs and polymorphs and to characterise them are described in chapters 5 and 6 of the book entitled "Polymorphism in Pharmaceutical SoUds” edited by Harry G Brittain (Marcel Dekker Inc., New York), pp. 183-278, 1999.
- the foUowing is a Ust of compounds that inhibit the efflux pump of different organisms.
- the list of compounds and organisms is not inclusive. There are compounds on this list that will inhibit the efflux pump of other organisms.
- Some specific efflux pump inhibitor compounds of the invention are:
- a microbe appropriate for the use of an efflux pump inhibitor are pathogenic bacterial species, such as Streptococcus pneumoniae, Streptococcus pyogenes, Pseudomonas aeruginosa, Escherischia coli, Staphylococcus aureus which can be intrinsically resistant to commonly used antibacterial agents. Exposing these bacteria to an efflux pump inhibitor can significantly slow the export of an antibacterial agent from the interior of the ceU or the export of siderophores. For instance, overexpression of the norA multidrug transporter has been reported for strains of S. aureus for fluoroquinolone resistance both in-vitro (Yoshida et.
- efflux pump inhibitors can decrease the virulence of the microbe, for example, by inhibiting the transport of factors important for pathogenicity.
- inhibition of an efflux pump in this bacterium inhibits the uptake of iron, which is important for pathogenicity.
- the mechanism of bacterial iron transport involves molecules called siderophores, which are synthesised and exported by bacterial ceUs via efflux pumps. These siderophores bind tightly to iron scavenged from the host, and are then taken up by the bacteria. In this way, the iron needed for bacterial metaboUsm is obtained, and an infection can be maintained.
- Streptococcus pneumoniae, Streptococcus pyogenes, Pseudomonas aeruginosa, Escherischia coli, Staphylococcus aureus strains wUl become susceptible to antibiotics that could not be used for treatment of the respective bacterial infections, or become more susceptible to antibiotics which do inhibit the respective bacterial growth.
- Streptococcus pneumoniae, Streptococcus pyogenes, Pseudomonas aeruginosa, Escherischia coli, Staphylococcus aureus strains will become more susceptible to antibiotics currently used for treatment of the respective bacterial infections.
- Virulence of Streptococcus pneumoniae, Streptococcus pyogenes, Pseudomonas aeruginosa, Escherischia coli, Staphylococcus aureus wiU be attenuated because the avaUabiUty of an essential siderephore bearing element wuT be hampered.
- microbes include, for example, bacteria, fungi, yeasts, and protozoa.
- the bacterium to be inhibited through the use of an efflux pump inhibitor can be from other bacterial groups or species, such bacterial groups of species including but not limited to one of the foUowing:
- Pseudomonas aeruginosa Pseudomonas fluorescens, Pseudomonas acidovorans, Pseudomonas alcaligenes, Pseudomonas putida, Stenotrophomonas maltophilia, Burkholderia capacia, Aeromonas hydrophilia, Escherichia coli, Citrobacter freundil, Salmonella tryphimurium, Salmonella typhi, Salmonella paratyphi, Salmonella enteritidis, Shigella dysenteriae, Shigella flexneri, Shigella sonnei, Enterobacter cloacae, Enterobacter aerogenes, Klebsiella pneumoniae, Klebsiella oxytoca, Serratia, marcescens, Francisella tularensis, Morganella morganii, Proteus mirabilis, Proteus vulgaris, Providencia alcalifaciens
- efflux pump refers to a protein assembly which exports substrate molecules from the cytoplasm or periplasm of a ceU, in an energy dependent fashion.
- an efflux pump wiU typicaUy be located in the cytoplasmic membrane of the cell (spanning the cytoplasmic membrane). In Gram-negative bacteria the pump may span the periplasmic space and there may also be portion of the efflux pump which spans the outer membrane.
- Certain efflux pumps wiU include a polypeptide which has at least 50% amino acid sequence similarity with a polypeptide which is part of the P.
- an "efflux pump inhibitor” is a compound which specifically interferes with the abflity of an efflux pump to export its normal substrate, or other compounds such as an antibiotic.
- the inhibitor may have intrinsic antimicrobial (e.g. antibacterial) activity of its own, but at least a significant portion of the relevant activity is due to the efflux pump inhibiting activity.
- compounds which inhibit the export or activity of efflux pumps which have a broad substrate range which includes antibacterial agents.
- Streptococcus pneumoniae-ty ⁇ >e efflux pump inhibitor refers to an efflux pump inhibitor which inhibits a Streptococcus pneumoniae-type efflux pump.
- Pseudomonas aeruginosa- type efflux pump inhibitor refers to an efflux pump inhibitor which inhibits a Pseudomonas aeruginosa-type efflux pump.
- Esscherischia coli-type efflux pump inhibitor refers to an efflux pump inhibitor which inhibits an Escherischia coli -type efflux pump.
- Staphylococcus aureus-type efflux pump inhibitor refers to an efflux pump inhibitor which inhibits a Staphylococcus aureus -type efflux pump.
- this invention provides a method for treating a microbial infection, e.g., a bacterial infection, in an animal by administering to an animal suffering from such an infection an efflux pump inhibitor as described above in an amount sufficient to reduce efflux pump activity.
- a microbial infection e.g., a bacterial infection
- the inhibitor is one which decreases the pathogenicity of the microbe.
- a decrease in pathogenicity can be obtained, for example, by interfering with essential bacterial element acquisition by inhibiting the transport of siderophores.
- the pathogenicity may also be reduced by reducing or eliminating the microbial products which cause tissue-damaging effects to the host. Other methods of reducing pathogenicity are, however, also within this aspect.
- the host is an animal and may be, for example, chickens and turkeys, and in certain preferred embodiments in a mammal, e.g. a human.
- the microbial infection may be due to bacteria, which may, for example, be any of the bacterial species indicated above, but specificaUy including Streptococcus pneumoniae, Pseudomonas aeruginosa, Escherischia coli, Staphylococcus aureus.
- this invention provides a method of treating an animal suffering from a microbial infection by administering to the animal an efflux pump inhibitor in an amount sufficient to reduce efflux pump activity.
- the efflux pump inhibitor in one which reduces the in vivo viabdity of a microbe involved in the infection.
- the infected animal can more readUy clear its body of the infection, or the microbes may even be lulled.
- the animal is a mammal.
- the microbe may be from one of a variety of pathogenic bacterial species, specificaUy including those Usted above.
- in vivo viabiUty refers to the abUity of a microbe, e.g., a bacterium, to survive or grow in a host, such as an animal. Therefore, an efflux pump inhibitor which reduces the in vivo viability of a microbe may stop the growth of the microbe and/or kill the microbe. Such efflux pump inhibitors, therefore, are antimicrobial agents.
- this invention includes a method for prophylactic treatment of an animal, e.g., a mammal.
- an efflux pump inhibitor which reduces the pathogenicity of a microbe is administered to a mammal at risk of a microbial infection, e.g., a bacterial infection.
- the invention provides a method for treating a microbial infection in an animal, specifically including in a mammal, by treating an animal suffering from such an infection with an antimicrobial agent and an efflux pump inhibitor which increases the susceptibUity of the microbe for that antimicrobial agent.
- a microbe involved in the infection can be treated using the antimicrobial agent in smaUer quantities, or can be treated with an antimicrobial agent which is not therapeuticaUy effective when used in the absence of the efflux pump inhibitor.
- this method of treatment is especially appropriate for the treatment of infections using an antimicrobial agent alone due to a need for high dosage levels (which can cause undesirable side effects), or due to lack of any cUnically effective antimicrobial agents.
- it is also appropriate for treating infections involving microbes which are susceptible to particular antimicrobial agents as a way to reduce the dosage of those particular agents. This can reduce the risk of side effects, but can also reduce the selection effect for highly resistant microbes resulting from the consistent high level use of a particular antimicrobial agent.
- the microbe is a bacterium, which may, for example, be from any of the groups or species indicated above. Also in particular embodiments various antibacterial agents can be used.
- an antibiotic of the above classes can be, for example, one of the foUowing: Beta-Lactam Antibiotics
- Amikacin arbekacin, butirosin, dibekacin, fortimicins, gentamicin, kanamycin, netilmicin, ribostanycin, sisomicin, spectinomycin, streptomycin, tobramycin, cUndamycin, Uncomycin.
- the term "susceptibiUty" refers to the sensitivity of the microbe for the presence of the antimicrobial agent So, to increase the susceptibiUty means that the microbe wUl be inhibited by a lower concentration of the antimicrobial agent in the medium surrounding the microbial ceUs. This is equivalent to saying that the microbe is more sensitive to the antimicrobial agent. In most cases the minimum inhibitory concentration (MIC) of that antimicrobial agent wiU have been reduced.
- MIC minimum inhibitory concentration
- the term “treating” refers to administering a pharmaceutical composition for prophylactic and/or therapeutic purposes.
- prophylactic treatment refers to treating an organism such as a human patient, who is not yet infected, but who is susceptible to, or otherwise at risk of, a particular infection.
- susceptible and risk do not refer to the status of organisms of that type generaUy, but rather refers to a significantly enhanced risk. Such risk may for example be due to a specific exposure to a particular potentially infective agent, to a generaUy weakened physical condition, or immune system deficiency.
- microbial infection refers to a disease state or some adverse condition ⁇ ), such as the presence of a pathogenic microorganism in a body fluid like blood, urine, cerebrospinal or organ tissue, which are otherwise sterile or free of pathogenic microorganisms.
- therapeutic treatment refers to administering treatment to a patient already suffering from an infection.
- treating is the administration to a mammal (either for therapeutic or prophylactic purposes) of therapeutically effective amounts of a potentiator and an antibacterial (or antimicrobial) agent in combination (either simultaneously or seriaUy).
- a potentiator refers to a compound such as an efflux pump inhibitor which has the abiUty to increase the concentration of existing antibiotics in a microbial cell.
- a therapeuticaUy effective amount is meant an amount of an efflux pump inhibitor, or amounts individually of an efflux pump inhibitor and an antimicrobial agent, as disclosed for this invention, which have a therapeutic effect, which generaUy refers to the inhibition to some extent of the normal metaboUsm of microbial cells causing or contributing to a microbial infection.
- the doses of efflux pump inhibitor and antimicrobial agent which are useful in combination as a treatment are therapeuticaUy effective amounts.
- a therapeuticaUy effective amount means those amounts of efflux pump inhibitor and antimicrobial agent which, when used in combination, produce the desired therapeutic effect as judged by cUnical trial results and/or model animal infection studies.
- the efflux pump inhibitor and antimicrobial agent are combined in predetermined proportions and thus a therapeutically effective amount would be an amount of the combination.
- This amount and the amount of the efflux pump inhibitor and antimicrobial agent individually can be routinely determined by one of skill in the art, and wiU vary, depending on several factors, such as the particular microbial strain involved and the particular efflux pump inhibitor and antimicrobial agent used. This amount can further depend upon the patient's height, weight, sex, age and medical history.
- a therapeuticaUy effective amount is that amount which would be effective if a microbial infection existed.
- a therapeutic effect reUeves, to some extent, one or more of the symptoms of the infection, and includes curing an infection.
- “Curing” means that the symptoms of active infections are eliminated, including the elimination of excessive numbers of viable microbes of those involved in the infection. However, certain long-term or permanent effects of the infection may exist even after a cure is obtained (such as extensive tissue damage).
- microbial infection refers to the invasion of the host mammal by pathogenic microbes. This includes the excessive growth of microbes which are normaUy present in or on the body of a mammal. More generaUy, a microbial infection can be any situation in which the presence of a microbial population(s) is damaging to a host mammal. Thus, a mammal is "suffering" from a microbial infection when excessive numbers of a microbial population are present in or on a mammal's body, or when the effects of the presence of a microbial population (s) is damaging the ceUs or other tissue of a mammal. SpecificaUy, this description applies to a bacterial infection.
- administering refers to a method of giving a dosage of an antimicrobial pharmaceutical composition to a mammal, where the method is, e.g., topical, oral, intravenous, intraperitoneal, or intramuscular.
- the preferred method of administration can vary depending on various factors e.g., the components of the pharmaceutical composition, the site of the potential or actual bacterial infection, the microbe involved, and the severity of an actual microbial infection.
- mammal is used in its usual biological sense. Thus, it specifically includes humans, cattle, horses, dogs, and cats, but also includes many other species.
- This invention also features a method of enhancing the antimicrobial activity of an antimicrobial agent against a microbe, in which such a microbe is contacted with an efflux pump inhibitor, e.g., a non-tetracycline specific efflux pump inhibitor, to an efflux pump in the cell, and an antibacterial agent.
- an efflux pump inhibitor e.g., a non-tetracycline specific efflux pump inhibitor
- the efflux pump inhibitor is a compound as described above.
- the microbe is a bacterium or a fungus, such as any of those described above;
- the antibacterial agent can be selected from a number of structural classes of antibiotics including, e.g., beta-lactams, glycopeptides, aminoglycosides, quinolones, tetracycUnes, rifamycins, coumermycins, macroUdes, and chloramphenicol.
- an antibiotic of the above classes can be as stated above.
- this invention provides pharmaceutical compositions effective for treatment of an infection of an animal, e.g., a mammal, by a microbe, such as a bacterium or a fungus.
- the composition includes a pharmaceutically acceptable carrier and an efflux pump inhibitor as described above.
- efflux pump inhibitors which are themselves effective antimicrobial agents, even in the absence of another antimicrobial agent (i.e., have intrinsic antimicrobial activity).
- pharmaceutical composition including such efflux pump inhibitors can be used either alone or in conjunction with another antimicrobial agent.
- the efflux pump inhibitors in pharmaceutical compositions of this aspect are efflux pump inhibitors which enhance the effectiveness of an antimicrobial agent other than the efflux pump inhibitor, so such compositions would generaUy be used in combination with such other antimicrobial agent.
- the invention also provides pharmaceutical compositions similarly effective for treatment of an infection of a mammal which include an efflux pump inhibitor and an antimicrobial agent.
- the invention provides antimicrobial formulations which include an antimicrobial agent, an efflux pump inhibitor, and a carrier.
- the antimicrobial agent is an antibacterial agent.
- a “carrier” or “excipient” is a compound or material used to facUitate administration of the compound, for example, to increase the solubUity of the compound.
- SoUd carriers include, e.g., starch, lactose, dicalcium phosphate, sucrose, and kaoUn.
- Liquid carriers include, e.g., sterUe water, saUne, buffers, non-ionic surfactants, and edible oils such as oil, peanut and sesame oils.
- various adjuvants such as are commonly used in the art may be included.
- the invention provides a method of suppressing growth of a microbe, e.g., a bacterium, expressing an efflux pump, e.g., a non-tetracycUne-specific efflux pump.
- a microbe e.g., a bacterium
- the method involves containing that bacterium with an efflux pump inhibitor, e.g., a non-tetracycline-specific efflux pump inhibitor, in the presence of a concentration of antibacterial agent below the MIC of the bacterium.
- an efflux pump inhibitor e.g., a non-tetracycline-specific efflux pump inhibitor
- This method is useful, for example, to prevent or cure contamination of a ceU culture by a bacterium possessing an efflux pump.
- the invention provides a method for reducing a population of a microbe, e.g., a bacterial strain, involving contacting the population with an efflux pump inhibitor which inhibits a component of an efflux pump expressed in the microbe in that population, which is essential for the growth of the microbe expressing that efflux pump.
- an efflux pump inhibitor which inhibits a component of an efflux pump expressed in the microbe in that population, which is essential for the growth of the microbe expressing that efflux pump.
- that component is cytoplasmic membrane component.
- efflux pump inhibitors may act in various ways, including, but not limited to, acting directly on the essential component, or acting to inhibit the expression of that component.
- an "essential component" of an efflux pump is one which is essential to the in vivo survival of the microbe, i.e., the survival in a host.
- this invention provides a method for enhancing growth of an animal by administering an efflux pump inhibitor to the animal, which inhibits an efflux pump expressed in a bacterial strain in the animal, and which inhibits the growth of the bacterial strain.
- an efflux pump inhibitor to the animal, which inhibits an efflux pump expressed in a bacterial strain in the animal, and which inhibits the growth of the bacterial strain.
- Such a growth enhancing effect may result from the reduced energy consumption by the bacteria, which increases the food energy available to the animal.
- This method is appropriate, for example, for use with cattle, swine, and fowl such as chickens and turkeys.
- the invention provides novel compounds having efflux pump activity. These compounds have chemical structures as described above. As indicated above, while the present invention is presently exemplified by activity against bacteria, compounds of the present invention also have activity against other microbes, for example against yeasts and/or other fungi. Thus, the above aspects also include embodiments in which described compounds are active or effective against such other microbes.
- the invention provides a method of making a pharmaceutical composition
- a method of making a pharmaceutical composition comprising the steps of identifying an efflux pump inhibitor having a chemical structure of the formula I; synthesizing said efflux pump inhibitor and preparing a pharmaceutical composition containing said efflux pump inhibitor.
- the efflux pump inhibitor may have the chemical structure as described above.
- the pharmaceutical composition may also contain one or more antimicrobial agents, e.g., as identified above, and one or more carriers, dUuents, and excipients.
- the efflux inhibitor compound is active against a microbe, e.g., a bacterium, as identified above.
- Identification of efflux pump inhibitors having structures as described for the present invention was performed using screening methods known to those skilled in the art of biological techniques and are described in detail below.
- the screening method based on inhibition of microbial growth in the presence of a subinhibitory concentration of an antibacterial agent which is normally effluxed by the test microbe and a concentration of a test compound was used for identifying some of the active compounds disclosed herein.
- inhibition of growth of the microbe is indicative that export of the antibacterial agent is inhibited by the test compound, and that the test compound is therefore an efflux pump inhibitor.
- the mode of action of the test compound so identified can then be confirmed as inhibiting active efflux.
- other screening methods for detecting efflux pump inhibitors can also be used.
- the inventors have screened a Ubrary of synthetic chemicals and identified several compounds that effectively inhibit the respective efflux pumps of Staphylococcus aureus 1199B NorA*, Streptococcus pneumoniae 3514, Pseudomonas aeruginosa 23587, Escherischia coli 2051. Some of these compounds were found to be also effective against presently unidentified multidrug transporters of other microorganisms.
- the library of compounds was obtained by synthesis according to methods as described in our copending applications PCT appUcation PCT/IN99/00016, US appUcations 09/566,875, 09/640,947, and 09/850,669 and by methodologies described in a later section below.
- Inhibitors of the bacterial efflux pumps are generaUy initially characterised in vitro. Those which show effective inhibition of the pump(s) and which show synergistic activity with antibiotics are selected for evaluation in vivo. Efficacy testing wUl be done using standard procedures. Primary efficacy evaluation may be done using the murine septicemia model (M.G. Bergeron, 1978, Scand. J. Infect. Dis. Suppl. 14:189-206; S.D. Davis, 1975, Antimicrob. Agents Chemother. 8:50-53). In this model a supra-lethal dose of bacteria is used to chaUenge the rodents. Treatment is initiated, varying either or both time(s) of treatment and dose of antibiotic. In these experiments both the antibiotic and the efflux pump inhibitor doses are varied. A positive result is indicated by significant increase in protection from the lethal infection by the combination of the potentiator (the efflux pump inhibitor) and the antibiotic versus the antibiotic alone.
- the efflux pump inhibitor the efflux pump inhibitor
- mice are infected with an appropriate titer of bacteria in the muscle of the hind thigh.
- Mice are either neutropenic (cyclophosphamide treated at 125 mg/kg on days -4, -2, and 0) or immunocompetent.
- the infecting dose is commonly 10 s - 10 6 colony forming units per animal.
- the proliferation (or death) of the bacteria within the thigh muscle is monitored over time. Effective combinations show greater activity than the antibiotic alone. Activity is defined as reduction in growth rate of the test bacteria in the murine tissue.
- the particular compound that is an efflux pump inhibitor can be administered to a patient either by itself, or in combination with an antimicrobial, e.g., antibacterial, agent, or in pharmaceutical compositions where it is mixed with a suitable carrier(s) or excipient(s) or diluent(s).
- a combination of an efflux pump inhibitor with an antimicrobial agent can be of at least two different types. In one, a quantity of an efflux pump inhibitor is combined with a quantity of an antimicrobial agent in a mixture, e.g., in a solution or powder mixture. In such mixtures, the relative quantities of the inhibitor and the antimicrobial agent may be varied as appropriate for the speciflc combination and expected treatment.
- an inhibitor and an antimicrobial agent can be covalently linked in such manner that the Unked molecules can be cleaved within the ceU.
- the term "in combination” can also refer to other possibilities, including serial administration of an inhibitor and other antimicrobial agent.
- an efflux pump inhibitor and/or another antimicrobial agent may be administered in pro-drug forms, i.e. the compound is administered in a form which is modified within the ceU to produce the functional form.
- a therapeuticaUy effective amount of an agent or agents such as these is administered.
- a therapeutically effective dose refers to that amount of the compound(s) that results in amelioration of symptoms or a prolongation of survival in a patient, and may include eUmination of a microbial infection.
- Toxicity and therapeutic efficacy of such compounds can be determined by standard pharmaceutical procedures in ceU cultures or experimental animals, e.g., for determining the LD 50 (the dose lethal to 50% of the population) and the ED 50 (the dose therapeutically effective in 50% of the population).
- the dose ratio between toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio LD 50/ ED 50 .
- Compounds which exhibit large therapeutic indices are preferred.
- the data obtained from these cell culture assays and animal studies can be used in formulating a range of dosage for use in human.
- the dosage of such compounds Ues preferably within a range of circulating concentrations that include the ED 50 with Uttle or no toxicity.
- the dosage may vary within this range depending upon the dosage and dosage form employed and the route of administration utiUsed. It is preferable that the therapeutic serum concentration of an efflux pump inhibitor should be in the range of 0.1-100 mcg/ml, more preferably 0.1 - 50 mcg/ml, even more preferably 0.1 - 20 mcg/ml, even more preferably 1.0- 50 mcg./ml or most preferably 1.0 - 20 mcg/ml.
- the therapeuticaUy effective dose can be estimated initiaUy from ceU culture assays.
- a dose can be formulated in animal models to achieve a circulating plasma concentration range that includes the IC 50 as determined in cell culture. Such information can be used to more accurately determine useful dosage in humans. Levels in plams may be measured, e.g. by HPLC.
- the efflux inhibitor in a pharmaceutical composition has a structure as shown by the generic structures described above.
- the exact formulation, route of administration and dosage can be chosen by the individual physician in view of the patients condition. (See e.g. Fingl etal., in The Pharmacological Basis of Therapeutics, 1975, Ch.l, p.l). It should be noted that the attending physician would know how and when to terminate, interrupt or adjust administration due to toxicity, or to organ dysfunctions. Conversely, the attending physician would also know to adjust treatment to higher levels if the cUnical response were not adequate, (precluding toxicity). The severity of the condition may, for example, be evaluated, in part, by standard prognostic evaluation methods. Further, the does and perhaps dose frequency wdl also vary according to the age, body weight and response of the individual patient. A programme comparable to that discussed above may be used in veterinary medicine.
- Such agents may be formulated and administered systemicaUy or locally.
- Techniques for formulation and administration may be found in Remington's Pharmaceutical Sciences, 19 th ed., Mack PubUshing Co., Easton, Pa. (1990). Suitable routes may include oral, rectal, transdermal, vaginal, transmucosal, or intestinal administration; parentral deUvery, including intramuscular, subcutaneous, intramedullary, injections, as well as intrathecal, direct intraventricular, intravenous, intraperitonial, intranesal, or intraocular injections just to name a few.
- compositions are prepared according to conventional procedures used by persons skilled in the art to make stable and effective compositions.
- an effective amount of the active compound or the active ingredient is any amount, which produces the desired results.
- the pharmaceutical compositions may contain the active compounds of the invention, their derivatives, salts or hydrates thereof, in a form to be administered alone, but generaUy in a form to be administered in admixture with a pharmaceutical carrier selected with regard to the intended route of administration and standard pharmaceutical practice.
- Suitable carriers which can be used are, for example, diluents or excipients such as fiUers, extenders, binders, emollients, wetting agents, disintegrants, surface active agents and lubricants which are usually employed to prepare such drugs depending on the type of dosage form.
- any suitable route of administration may be employed for providing the patient with an effective dosage of the compound of the invention, their derivatives, salts or hydrates thereof.
- oral, rectal, parenteral (subcutaneous, intramuscular, intravenous), transdermal, topical and Uke forms of administration may be employed.
- Dosage forms include (solutions, suspensions, etc) tablets, piUs, powders, troches, dispersions, suspensions, emulsions, solutions, capsules, injectable preparations, patches, ointments, creams, lotions, shampoos and the like.
- compositions of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets, or tablets, or aerosol sprays, each containing a predetermined amount of the active ingredient, as a powder or granules, or as a solution or a suspension in an aqueous liquid, a non-aqueous liquid, an oil-in-water emulsion, or a water-in-oU liquid emulsion.
- Such compositions may be prepared by any of the methods of pharmacy, but aU methods include the step of bringing into association the active ingredient with the carrier which constitutes one or more necessary ingredients.
- the compositions are prepared by uniformly and intimately admixing the active ingredient with Uquid carriers or finely divided solid carriers or both, and then, if necessary, shaping the product into the desired presentation.
- compositions of the present invention include compositions such as suspensions, solutions, elixirs, aerosols, and solid dosage forms.
- Carriers as described in general above are commonly used in the case of oral soUd preparations (such as powders, capsules and tablets), with the oral soUd preparations being preferred over the oral liquid preparations.
- the most preferred oral soUd preparation is tablets.
- tablets and capsules represent the most advantageous oral dosage unit form, in which case solid pharmaceutical carriers are employed.
- suitable carriers include excipients such as lactose, white sugar, sodium chloride, glucose solution, urea, starch, calcium carbonate, kaolin, crystalUne ceUulose and sUicic acid, binders such as water, ethanol, propanol, simple syrup, glucose, starch solution, gelatine solution, carboxymethyl cellulose, shellac, methyl ceUulose, potassium phosphate and polyvinyl pyrroUdone, disintegrants such as dried starch, sodium alginate, agar powder, laminaria powder, sodium hydrogen carbonate, calcium carbonate, Tween (fatty acid ester of polyoxyethylenesorbitan), sodium lauryl sulfate, stearic acid monoglyceride, starch, and lactose, disintegration inhibitors such as white sugar, stearic acid glyceryl ester, cacao butter and hydrogenated
- the tablet if desired, can be coated, and made into sugar-coated tablets, gelatine-coated tablets, enteric-coated tablets, film-coated tablets, or tablets comprising two or more layers. If desired, tablets may be coated by standard aqueous or nonaqueous techniques.
- a wide variety of conventional carriers known in the art can be used.
- suitable carriers are excipients such as glucose, lactose, starch, cacao butter, hardened vegetable oils, kaoUn and talc, binders such as gum arabic powder, tragacanth powder, gelatin, and ethanol, and disintegrants such as laminaria and agar.
- a wide variety of carriers known in the art can be used.
- suitable carriers include polyethylene glycol, cacao butter, higher alcohols, gelatine, and semi-synthetic glycerides.
- a second preferred method of administration is parenteraUy for intramuscular, intravenous or subcutaneous administration.
- a third preferred route of administration is topicaUy, for which creams, ointments, shampoos, lotions, dusting powders and the like are well suited.
- the compounds of the present invention may also be administered by controlled release means and/or delivery devices such as those described in U S Patent Nos. 3,845,770; 3,916,899; 3,536,809; 3,598,123 and 4,008,719; the disclosures of which are hereby incorporated by reference.
- aU diluents customarUy used in the art can be used.
- suitable diluents are water, ethyl alcohol, polypropylene glycol, ethoxylated isostearyl alcohol, polyoxyethylene sorbitol, and sorbitan esters.
- Sodium chloride, glucose or glycerol may be incorporated into a therapeutic agent.
- the antimicrobial pharmaceutical composition may further contain ordinary dissolving aids, buffers, pain-aUeviating agents, and preservatives, and optionaUy colouring agents, perfumes, flavours, sweeteners, and other drugs.
- viscous to semi-soUd or soUd forms comprising a carrier compatible with topical application and having a dynamic viscosity preferably greater than water.
- Suitable formulations include but are not Umited to solutions, suspensions, emulsions, creams, ointments, powders, Uniments, salves, aerosols, etc., which are, if desired, steriUzed or mixed with auxiUary agents, e.g. preservatives, antioxidants, stabUizers, wetting agents, buffers or salts for influencing osmotic pressure, etc.
- auxiUary agents e.g. preservatives, antioxidants, stabUizers, wetting agents, buffers or salts for influencing osmotic pressure, etc.
- sprayable aerosol preparations wherein the active ingredient preferably in combination with a soUd or liquid inert carrier material.
- the compounds of the present invention may be readily prepared in accordance with the foUowing synthesis schemes, as iUustrated in the specific examples provided.
- those skilled in the art wUl recognise that other synthetic pathways for forming the compounds of this invention can be utilised, and that the following is provided merely by way of example and is not limiting to the present invention.
- various protecting and deprotecting strategies wiU be employed which are standard in the art (see e.g., "Protective Groups in Organic Synthesis", by Green and Wuts).
- protecting and deprotecting strategies wiU be employed which are standard in the art (see e.g., "Protective Groups in Organic Synthesis", by Green and Wuts).
- Those skilled in the art wuT recognise that the selection of any particular protecting group (e.g. amine, hydroxy and carboxyl protecting groups) wiU depend on the stabUity of the protected moiety with regard to the subsequent reaction conditions and wUl understand the appropriate selection.
- R,, R4 a , R s , Re, X and Y are as hereinbefore described is treated with an appropriate amine of the formula R 9 R ⁇ 0 NH, where R 9 and R ⁇ 0 have the meanings hereinbefore described in an organic solvent such as acetone, alcohol, acetonitrUe, dimethyl sulphoxide, N,N- dimethylformamide preferably acetonitrile or dimethyl sulphoxide, optionaUy in the presence of a base such as triethylamine, pyridine, l,5-diazabicyclo[4.3.0]non-5-ene (DBN), diazabicyclo[5.4.0]undee-7-ene (DBU) preferably triethylamine at 50°C - 120°C, preferably 70°C - 90°C for 4- 24 hr.
- a base such as triethylamine, pyridine, l,5-diazabicyclo[4.3.0
- the product obtained is hydrolysed by aqueous alkali preferably sodium hydroxide or a base preferably triethylamine in solution in a solvent such as ethanol.
- the compounds I of the invention which are esters at a carboxylic acid group may be prepared by treating the free acid of compounds of formula I in solution in an appropriate solvent, preferably N,N-dimethylformamide, with the corresponding halo compound, preferably chloro or bromo-compound, in the presence of a base, preferably anhydrous potassium carbonate, at an elevated temperature, preferably 50°C for an extended period of time, preferably 6 hours.
- an appropriate solvent preferably N,N-dimethylformamide
- halo compound preferably chloro or bromo-compound
- the compounds of the invention which are esters at a carboxylic acid group may be prepared by treating the free acid of compound of formula I in solution in an appropriate solvent, preferably N,N-dimethylacetamide, with the corresponding hydroxy compound, in the presence of a base, preferably triethylamine, in presence of a catalyst, preferably 4-N,N- dimethylaminopyridine, and in the presence of a dehydrating agent, preferably N,N- dicyclohexylcarbodimide at an elevated temperature, preferably 100°C for an extended period of time, preferably 24 hours.
- an appropriate solvent preferably N,N-dimethylacetamide
- a base preferably triethylamine
- a catalyst preferably 4-N,N- dimethylaminopyridine
- a dehydrating agent preferably N,N- dicyclohexylcarbodimide at an elevated temperature, preferably 100°C for an extended period of time, preferably 24 hours.
- the compounds of formula I of the invention which are amides at a carboxylic acid groups may be prepared by coupling the free acid of compound of formula I with ammonia or an appropriate amine or an amino acid appropriately protected at the acid functionality of the amino acids with a protecting group.
- the -COOH protecting groups for amino acids are known in the art. Examples of suitable -COOH protecting groups for amino acids are methyl, ethyl, t-butyl and benzyl groups.
- the -COOH protecting group is removed by hydrolysis or by hydrogenation.
- the coupling of a -COOH group of compound of formula I with the amino group of the amino acid is also known in the art.
- the reaction may be conducted with or without a solvent at a range of temperatures in the presence of a coupling agent.
- the compounds of the invention which are amides at an NH2 or an NH group are prepared by coupling the free amino group of a compound of formula I or by coupling the free NH bearing compound of formula I with an appropriate acylating agent such as an acyl anhydride or an acyl chloride in the presence of a condensing agent such as a base e.g. triethylamine or aqueous sodium hydroxide optionaUy in the presence of a solvent such as
- N,N-dimethyl acetamide at an elevated temperature of 50°C - 100°C for an extended period of time upto 24 hours.
- a protecting group on the NH2 group or NH group which is desired to remain unreactive For compound that contain two NH2 groups, two NH groups or one NH2 and one NH group, it may be necessary when so desired to use a protecting group on the NH2 group or NH group which is desired to remain unreactive.
- Protecting groups for NH2 and NH groups are weU known to those skiUed in the art.
- the compounds of the invention which are esters of a free hydroxy group may be prepared by treating the free hydroxy compound of formula I with an organic acid, an organic dibasic acid or appropriate N-protected amino acid or polypeptide as defined above.
- Nitrogen protecting groups are known in the art Examples of suitable nitrogen protecting groups are
- C C 6 acyl C 2 -C 6 alkoxycarbonyl optionally substituted benzyloxycarbonyl, aryloxycarbonyl, sUyl, trityl, tetrahydropyranyl, vinyloxycarbonyl, O-nitrophenylsulfonyl, diphenylphosphinyl, p-toluenesulfonyl, and benzyl.
- the nitrogen protecting group is removed by methods known in the art such as hydrogenation or hydrolysis.
- the ester forming reaction may be conducted with or without a solvent at a range of temperatures in the presence of a suitable condensing agent, known to those skilled in the art.
- the compounds of the invention which are alkyl ethers of a free hydroxy group may be prepared by treating the compound bearing the free hydroxy group with an alkyl halide in an organic solvent in the presence of a base or a condensing agent at temperatures upto the boiUng point of the solvent for a period of time upto 24 hours, by methods known to those skilled in the art.
- the compounds of the invention which are mono or dialkyl derivatives of a free amino group may be prepared by treating the compound bearing the free amino group with appropritae molar amounts of an alkylhalide in an organic solvent optionally in the presence of a base or a condensing agent at temperatures upto the boding point of the solvent for a period of time upto 24 hours, by methods known to those skilled in the art h) General Method for making aminoacid esters at the 8-(4'-hydroxypiperidine substituent of
- Trifluoroacetic acid (10 ml) was added to the product obtained in the previous step. After 30 mins at room temperature, the acid was evaporated and the trifluoroacetate salt was precipitated by addition of ether. If need be the product could be purified by high pressure
- the mixture was acidified, extracted with ethylacetate, washed with brine , dried over sodiumsulphate and evaporated to provide the title compound. If need be the product may be purified by high pressure Uquid chromatography on a C8 or C18 column. OptionaUy, dissolving the acid in water with one equivalent sodiumbicarbonate foUowed by freeze drying provided the sodium salt of the title compound.
- the pharmaceutically acceptable acid addition salts of compounds I are prepared in a conventional manner by treating a solution or suspension of the free base I with about one chemical equivalent of a pharmaceuticaUy acceptable acid. Conventional concentration and recrystalisation techniques are employed in isolating the salts.
- IUustrative of suitable acids are hydrochloric, hydrobromic, hydroiodic, sulphuric, sulfamic, sulfonic, phosphoric, acetic, lactic, succinic, oxaUc, maleic, fumaric, malic, tartaric, citric, ascorbic, gluconic, benzoic, cinnamic, methanesulfonic and p-toluenesulfonic acid.
- the pharmaceutically acceptable cationic salts of compounds of formula I may be prepared by conventional methods from the corresponding acids e.g. by reaction with about one equimolar amount of a base.
- suitable cationic salts are those of alkaU metals such as lithium, sodium or potassium, alkaline earth metals such as magnesium or calcium or ammonium or organic amines such diethanolamine, N-methylglucamine, guanidine or heterocyclic amines such as choUne, piperidine, N-methyl-4-hydroxypiperidine, hydroxyethylpyrroUdine, hydroxyethylpiperidine, morpholine, hydroxyethylmorphoUne, piperazine, N-methyl piperazine and the Uke or basic amino acids such as optically pure or racemic isomers of arginine, lysine, histidine, tryptophan and the like.
- Example 1 l-Ethyl-6-fluoro-l, 4-dihvdro -7-fl 2 f , 3', 4'-tetrahvdroisoquinoUn-2-yl)-4-oxo-quinoline-3- carboxylic acid.
- Example 12 l-Cyclopropyl-6-fluoro-l.4-dihvdro -7- ⁇ 4'-(N-dimethylamino) piperidin-l-yl)-4-oxo-quinoline-3- carboxyUc acid.
- Example 13 l-Cvclopropyl-6-fluoro-l, 4-dihvdro- -7-(3', 5'-dimethyl piperidin-l-ylV4-oxo-quinoUne-3- carboxylic acid.
- Example 14 l-Cvclopropyl-6-fluoro-l, 4-dihvdro -7-(4'-hydroxy-3 , , 5 , -dimethylpiperidin-l-ylV4-oxo-quinoUne- 3-carboxyUc acid. It was prepared in a similar manner as described in example 9 where 4-hydroxy-3,5- dimethylpiperidine was used in place of 3-acetamido-5-methyl pyrroUdine. Yield 38 %, m.p 178- 80 °C, C 20 H 23 FN 2 O 4 , m z 375 (M+l).
- Example 15 l-Cvclopropyl-6-fluoro-l, 4-dihvdro- -7-(3', 4', 5'-trimethyl piperazin-l-yl)-4-oxo-quinoline-3- carboxylic acid.
- Example 16 l-Cvclopropyl-6-fluoro-l, 4-dihvdro- 7-(3', 5 f -dimethyl-4 , -ethyl piperazin-l-yl)-4-oxo-quinoline-3- carboxylic acid.
- Example 18 l-Cvclopropyl-6-fluoro-l, 4-dihvdro-5-methyl- 7-(3 f , 3'-dimethylpiperazin -l-vD-4-oxo-quinoUne- 3-carboxylic acid.
- Example 20 l-Cyclopropyl-6-fluoro-8-methoxy-l, 4-dihvdro -7-(4'-hvdroxy-3 '-isobutylpiperidin-l-vD-4-oxo- quinoUne-3-carboxyUc acid.
- Example 21 l-Cvclopropyl-6-fluoro-8-methoxy-l, 4-dihvdro -7-(4'-hvdroxy-3', 3 f -dimethvIpiperidin-l-yl)-4- oxo-quinoUne-3-carboxyUc acid.
- Example 22 l-Cvclopropyl-6-fluoro-8-methoxy-l, 4-dihvdro ⁇ -H'-hvdroxy-S', 5 , -dimethylpiperidin-l-yl)-4- oxo-quinoline-3-carboxyUc acid.
- Example 23 l-Cvclopropyl-6-fluoro-8-methoxy-l, 4-dihvdro -7-(3 , -methylpiperazin-l-yl)-4-oxo-quinoline-3- carboxylic acid. It was prepared in a similar manner as described in example 19 where 3-methyl piperazine was used in place of 4-(N-dimethylamino)-3-methylpiperidine. Yield (48 %), m.p 260-61 °C, C ⁇ 9 H 22 FN 3 0 4 , m/z 375 (M+l).
- Example 1 l-Cvclopropyl-6-fluoro-l, 4-dihvdro-5-methyl- 7-(4 , -methoxypiperidin -l-yl)-4-oxo-quinoline-3- carboxylic acid.
- Example 12 l-Ethyl-6, 8-difluoro-l, 4-dihvdro -7-(3'-5'-dimethylpiperazin-l-yl)-4-oxo-quinoline-3-carboxyUc acid.
- Example 13 l-Cyclopropyl-6-fluoro-l, 4-dihvdro-7- (4'-ethyl-3 , -methylpiperazin-l-yl)-4-oxo-quinoline-3- carboxylic acid.
- Example 14 l-Cyclopropyl-6-fluoro-l.4-dihvdro- -7-(3 f -5'-dimethyl-4 , -ethylpiperazin-l-ylV4-oxo-quinoUne-3- carboxylic acid.
- Example 19 l-(2', 4 , -Difluorophenyl)-6-fluoro-l, 4-dihvdro-7- (piperidin-4 , -ylaminoV4-oxo-naphthyridine-3- carboxylic acid. Prepared according to example 49 in the list of compounds for the NorA pump.
- Example 20 l-Cvclopropyl-6-fluoro-l, 4-dihvdro -7-(4 , -amino-3'-ethylpiperidin-l-yl)-4-oxo-naphthyridine-3- carboxylic acid.
- Trifluoroacetic acid (10 ml) was added to the derivative obtained, stirred for 30 mins at room temp., the acid was evaporated and the trifluoroacetate salt was precipitated by addition of ether. Purified by RP-HPLC on a C18 column. Dissolved the trifluoroacetate salt in 0.1 N hydrochloride acid and freeze-dried provided the titled salt. Yield 75 %, m.p (hygroscopic) °C, C 23 H 26 FN 3 0 7 , m/z 543 (M+l)
- the compound was purified by prep-HPLC using water-acetonitrile-0.1% trifluoroacetic acid system to get after freeze drying a white powder as trifluoro acetate salt. Yield 74 %, m.p 80-82 °C, C 30 H 34 F 2 N 4 O 5 , m/z 467.5 (M+l).
- SoUd separated was filtered, dried and purified on sUica column to furnish titled product. Yield 66 %, m.p 150-52 °C, C 20 H 23 FN 2 O 4 , m/z 375 (M+l).
- Example 23 l-(2,4-Difluorophenyl) -6-fluoro-l, 4-dihvdro-7- ((loc, 5oc, 6oc)-6-amino-N-benzyl-3-azabicvclo [3.1.01 hex-6-yl - 4-oxo-l,8-naphthyridine-3-carboxylic acid.
- Example 30 ( S ) -f-V9-Fluoro-6.7-dihvdro-8- (3 , . 3 , -dimethyl-4 , -ethylaminopiperidin-l-vn-5-methyl-l-oxo-lH, 5H-benzo fi.i[quinoUzine-2-carboxyUc acid.
- FIC Fractional Inhibition Concentration
- the assay involves broth microdilution method performed as recommended by the National Committee for CUnical Laboratory Standards (NCCLS) 1997 (Methods for Dilution of Antimicrobial Susceptibility Tests for Bacteria that Grow AerobicaUy, Fourth edition; Approved
- test organism used for the assay is Staphylococcus aureus 1199 B NorA + .
- DMSO dimethyl sulfoxide
- the checkerboard assay is performed in microtiter plates. Ciprofloxacin is diluted in the X-axis, each column containing a single concentration of ciprofloxacin. The efflux pump inhibitor is diluted in the Y-axis, each row containing an equal concentration of efflux pump inhibitor. The result of these manipulations is that each well of the microtiter plate contains a unique combination of concentrations of the two agents. Bacterial inoculum is added at 5 x 10 s CFU/ml. Microtiter plates are incubated at 35°C for 20 hours. Reading is done by visibly noting turbidity and confirmed using a microtiter plate reader (Molecular Devices). The FIC index is calculated as
- (A) is the concentration of the experimental efflux pump inhibitor in the weU that is the lowest inhibitory concentration in the Y-axis.
- (MIC A ) is the MIC of Staphylococcus aureus to experimental efflux pump inhibitor alone.
- FIC A is the fractional inhibitory concentration of an experimental efflux pump inhibitor.
- (B) is the concentration of the ciprofloxacin in the weU that is the lowest inhibitory concentration in the X-axis.
- FIC B is the fractional inhibitory concentration of ciprofloxacin.
- Pseudomonas aeruginosa 23587 is used.
- Pseudomonas aeruginosa 23587 is a cUnical isolate containing multiple efflux pumps which can be mexAB, mexCD, mexEF but not Umited to mex classification only.
- Pseudomonas aeruginosa are grown in tryptic soya broth for 20 hours at 35°C.
- Two antibiotic assay plates are prepared in Mueller Hinton Agar using above inoculum. The bacterial count in a plate is 1 x 10 6 ceUs/ml.
- levofloxacin is incorporated at 5 mcg/ml, whUe in the control plate an equivalent amount of water is added.
- An experimental efflux pump inhibitor is evaluated by adding a series of concentrations of the test compound ranging from 0.025 meg to 200 mcg./well in agar plate. The plates are incubated at 35°C for 20 hours and the zone of inhibition was recorded in mm (table 2). The difference in the diameter of zone of inhibition was calculated by subtracting from the diameter of the zone of inhibition in the levofloxacin containing plate the diameter of the zone of inhibition in the water containing control plate.
- E.coli 2051 The activity of an efflux pump inhibitor of E.coli 2051 was also assessed using an antibiotic diffusion assay.
- Exoli 2051 is used.
- E.coli 2051 is a clinical isolate containing multiple efflux pumps.
- E.coli 2051 are grown in tryptic soya broth for 20 hours at 35°C.
- Two antibiotic assay plates are prepared in Mueller Hinton Agar using above inoculum. The bacterial count in a plate is 1 x 10 6 cells/ml. In one of the plates, levofloxacin is incorporated at 15 mcg/ml, whUe in the control plate an equivalent amount of water is added.
- An experimental efflux pump inhibitor is evaluated by adding a series of concentrations of the test compound ranging from 0.025 meg to 200 mcg./well in agar plate. The plates are incubated at 35°C for 20 hours and the zone of inhibition was recorded in mm (table 3) . The difference in the diameter of zone of inhibition was calculated by subtracting from the zone of inhibition in the levofloxacin containing plate the diameter of the zone of inhibition in the control plate.
- Streptococcus pneumoniae 3514 (Mef+) is used which was obtained from the Centre for Disease Control (CDC), Atlanta. A simUar 2 plate assay method as described above is used with small modifications. Streptococcus pneumoniae 3514 (Mef+) is grown in Columbia blood agar for 18 hours at 25°C. The growth was suspended in sterile todd hewitt broth and CFU were adjusted using macfarland's standard to 1 x 10 s CFU/ml. 0.5 ml of this is added to the MueUer Hinton agar supplemented with 5% sheep blood, final bacterial count was 10 6 ceUs/ml.
- Streptococcus pyogenus 26-00 is used which was obtained from the Centre for Disease Control (CDC), Atlanta. A similar 2 plate assay method as described above is used with small modifications. Streptococcus pyogenus 26-00 (Mef+) is grown in Columbia blood agar for 18 hours at 25°C. The growth was suspended in sterUe todd hewitt broth and CFU were adjusted using macfarland's standard to 1 x 10 s CFU/ml. 0.5 ml of this is added to the Mueller Hinton agar supplemented with 5% sheep blood, final bacterial count was 10 6 ceUs/ml.
- *Z the difference of the diameter of the zone of inhibition in the levofloxacin containing plate minus the diameter of the zone of inhibition in the water containing control plate. * S-isomer; ** R-isomer; Absence of an asterisk indicates that the substance is a racemic compound.
- Z the difference of the diameter of the zone of inhibition in the levofloxacin containing plate minus the diameter of the zone of inhibition in the water containing control plate.
- *Z the difference of the diameter of the zone of inhibition in the levofloxacin containing plate minus the diameter of the zone of inhibition in the water containing control plate. * S-isomer; ** R-isomer; Absence of an asterisk indicates that the substance is a racemic compound.
- *Z the difference of the diameter of the zone of inhibition in the levofloxacin containing plate minus the diameter of the zone of inhibition in the water containing control plate. * S-isomer; ** R-isomer; Absence of an asterisk indicates that the substance is a racemic compound.
Abstract
Description



























Claims

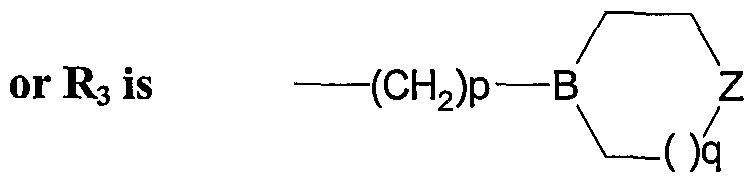








































Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002417799A CA2417799A1 (en) | 2000-08-01 | 2001-07-31 | Inhibitors of cellular efflux pumps of microbes |
| AU2001280091A AU2001280091A1 (en) | 2000-08-01 | 2001-07-31 | Inhibitors of cellular efflux pumps of microbes |
| EP01958373A EP1305048A2 (en) | 2000-08-01 | 2001-07-31 | Inhibitors of cellular efflux pumps of microbes |
Applications Claiming Priority (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US22220100P | 2000-08-01 | 2000-08-01 | |
| US60/222,201 | 2000-08-01 | ||
| US09/640,947 | 2000-08-17 | ||
| US09/640,947 US6750224B1 (en) | 1999-05-07 | 2000-08-17 | Antibacterial optically pure benzoquinolizine carboxylic acids, processes, compositions and methods of treatment |
| IN0000111 | 2000-11-22 | ||
| INPCT/IN00/00111 | 2000-11-22 | ||
| US28629101P | 2001-04-25 | 2001-04-25 | |
| US60/286,291 | 2001-04-25 | ||
| US09/850,669 | 2001-05-07 | ||
| US09/850,669 US6608078B2 (en) | 2000-05-08 | 2001-05-07 | Antibacterial chiral 8-(substituted piperidino)-benzo [i,j] quinolizines, processes, compositions and methods of treatment |
| INPCT/IN01/00100 | 2001-05-08 | ||
| PCT/IN2001/000100 WO2001085728A2 (en) | 2000-05-08 | 2001-05-08 | Antibacterial chiral 8-(substituted piperidino)-benzo [i, j] quinolizines, processes, compositions and methods of treatment |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2002009758A2 true WO2002009758A2 (en) | 2002-02-07 |
| WO2002009758A3 WO2002009758A3 (en) | 2002-12-27 |
Family
ID=27517699
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2001/000139 WO2002009758A2 (en) | 2000-08-01 | 2001-07-31 | Inhibitors of cellular efflux pumps of microbes |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP1305048A2 (en) |
| AU (1) | AU2001280091A1 (en) |
| CA (1) | CA2417799A1 (en) |
| WO (1) | WO2002009758A2 (en) |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002085886A2 (en) * | 2001-04-25 | 2002-10-31 | Wockhardt Limited | Chiral, broad-spectrum antibacterial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation and compositions |
| WO2003050107A1 (en) * | 2001-12-13 | 2003-06-19 | Wockhardt Limited | New generation triple-targeting, chiral, broad-spectrum antimicrobial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation, compositions and use as medicaments |
| US6608078B2 (en) | 2000-05-08 | 2003-08-19 | Wockhardt Limited | Antibacterial chiral 8-(substituted piperidino)-benzo [i,j] quinolizines, processes, compositions and methods of treatment |
| WO2003099815A1 (en) * | 2002-05-28 | 2003-12-04 | Wockhardt Limited | Crystalline fluoroquinolone arginine salt form |
| GB2403653A (en) * | 2003-07-10 | 2005-01-12 | Secr Defence | Use of fluoroquinone antibiotics for the treatment of plague |
| WO2005023805A1 (en) * | 2003-09-04 | 2005-03-17 | Wockhardt Limited | Benzoquinolizine-2-carboxylic acid arginine salt tetrahydrate |
| WO2005026145A2 (en) * | 2003-09-12 | 2005-03-24 | Warner-Lambert Company Llc | Quinolone antibacterial agents |
| US6878713B2 (en) | 2001-04-25 | 2005-04-12 | Wockhardt Limited | Generation triple-targeting, chiral, broad-spectrum antimicrobial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation, compositions and use as medicaments |
| WO2005070941A1 (en) * | 2004-01-13 | 2005-08-04 | Cumbre Pharmaceuticals Inc. | Rifamycin imino derivatives effective against drug-resistant microbes |
| WO2005089738A2 (en) * | 2004-03-17 | 2005-09-29 | Mpex Pharmaceuticals, Inc. | Use and administration of bacterial efflux pump inhibitors |
| WO2005070940A3 (en) * | 2004-01-13 | 2005-09-29 | Cumbre Inc | Rifamycin derivatives effective against drug-resistant microbes |
| US7098219B2 (en) | 2000-08-01 | 2006-08-29 | Wockhart Limited | Inhibitors of cellular efflux pumps of microbes |
| WO2007106537A3 (en) * | 2006-03-13 | 2007-11-29 | Activx Biosciences Inc | Aminoquinolones as gsk-3 inhibitors |
| WO2008019292A2 (en) * | 2006-08-04 | 2008-02-14 | Trustees Of Boston University | Compositions and methods for potentiating antibiotic activity |
| WO2007102061A3 (en) * | 2006-03-07 | 2009-04-23 | Wockhardt Ltd | Prodrugs of benzoquinolizine-2-carboxylic acid |
| US7569563B2 (en) | 2005-06-30 | 2009-08-04 | Aicuris Gmbh & Co. Hk | Substituted quinolones II |
| EP1918289A3 (en) * | 2002-05-28 | 2009-08-19 | Wockhardt Limited | Crystalline nadifloxacin arginine salt form |
| US7829543B2 (en) | 2003-01-07 | 2010-11-09 | Paratek Pharmaceuticals, Inc. | Substituted polyamines as inhibitors of bacterial efflux pumps |
| US7867992B2 (en) | 2004-07-21 | 2011-01-11 | Aicuris Gmbh & Co. Kg | Substituted quinolones |
| US7902227B2 (en) | 2007-07-27 | 2011-03-08 | Janssen Pharmaceutica Nv. | C-7 isoxazolinyl quinolone / naphthyridine derivatives useful as antibacterial agents |
| US7977349B2 (en) | 2006-02-09 | 2011-07-12 | Aicuris Gmbh & Co. Kg | Substituted quinolones III |
| US7994225B2 (en) | 2004-03-17 | 2011-08-09 | Rempex Pharmaceuticals, Inc. | Bacterial efflux pump inhibitors for the treatment of ophthalmic and otic infections |
| WO2011101710A1 (en) * | 2010-02-16 | 2011-08-25 | Wockhardt Research Centre | Efflux pump inhibitors |
| US8071591B2 (en) | 2009-03-11 | 2011-12-06 | Kyorin Pharmaceutical Co., Ltd. | 7-cycloalkylaminoquinolones as GSK-3 inhibitors |
| CN102351880A (en) * | 2007-09-11 | 2012-02-15 | 杏林制药株式会社 | Cyanoaminoquinolones and tetrazoloaminoquinolones as GSK-3 inhibitors |
| US8476261B2 (en) | 2007-09-12 | 2013-07-02 | Kyorin Pharmaceutical Co., Ltd. | Spirocyclic aminoquinolones as GSK-3 inhibitors |
| CN103183684A (en) * | 2011-12-29 | 2013-07-03 | 杭州师范大学 | Quinolone lactone compound, and preparation method and application thereof |
| EP2751083A4 (en) * | 2011-08-31 | 2015-06-10 | Otsuka Pharma Co Ltd | Quinolone compound |
| US9155792B2 (en) | 2006-02-13 | 2015-10-13 | Trustees Of Boston University | RecA inhibitors with antibiotic activity, compositions and methods of use |
| CN109486739A (en) * | 2018-11-23 | 2019-03-19 | 上海海洋大学 | A method of induction vibrio parahemolyticus generates levofloxacin resistance |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0300735A1 (en) * | 1987-07-20 | 1989-01-25 | CHINOIN Gyogyszer és Vegyészeti Termékek Gyára RT. | Antimicrobial pharmaceutical composition |
| WO1997024128A1 (en) * | 1995-12-27 | 1997-07-10 | Bayer Corporation | Otic microbial combinations |
-
2001
- 2001-07-31 EP EP01958373A patent/EP1305048A2/en not_active Withdrawn
- 2001-07-31 CA CA002417799A patent/CA2417799A1/en not_active Abandoned
- 2001-07-31 WO PCT/IN2001/000139 patent/WO2002009758A2/en not_active Application Discontinuation
- 2001-07-31 AU AU2001280091A patent/AU2001280091A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0300735A1 (en) * | 1987-07-20 | 1989-01-25 | CHINOIN Gyogyszer és Vegyészeti Termékek Gyára RT. | Antimicrobial pharmaceutical composition |
| WO1997024128A1 (en) * | 1995-12-27 | 1997-07-10 | Bayer Corporation | Otic microbial combinations |
Non-Patent Citations (5)
| Title |
|---|
| A.M.DHOPLE, M.A.IBANEZ: "In vivo susceptibility of mycobacterium leprae to ofloxacin either singly or in combination with rifampicin and rifabutin" ARZNEIMITTEL-FORSCHUNG, vol. 44, no. 4, 1994, pages 563-565, XP001108849 * |
| E.M.MATEU-DE-ANTONIO, M.MARTIN: "In vitro efficacy of several antimicrobial combinations against Brucella canis and Brucella melitensis strains isolated from dogs" VETERINARY MICROBIOLOGY, vol. 45, no. 1, 1995, pages 1-10, XP001112917 * |
| H.C.NEU, N.X.CHIN: "In vitro activity of fleroxacin in combination with other antimicrobial agents" AMERICAN JOURNAL OF MEDICINE, vol. 94, no. 3A, 1993, pages 9S-16S, XP008008698 * |
| N.X.CHIN, H.C.NEU: "Combination of ofloxacin and other antimicrobial agents" JOURNAL OF CHEMOTHERAPY (FLORENCE), vol. 2, no. 6, 1990, pages 343-347, XP008008692 * |
| P.VAN DER AUWERA E.A.: "Comparative in vitro activity of CI934, a new fluoroquinolone, alone and in combination with coumermycin, against gram-positive bacteria" DRUGS UNDER EXPERIMENTAL AND CLINICAL RESEARCH, vol. 13, no. 3, 1987, pages 125-132, XP008008697 * |
Cited By (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6608078B2 (en) | 2000-05-08 | 2003-08-19 | Wockhardt Limited | Antibacterial chiral 8-(substituted piperidino)-benzo [i,j] quinolizines, processes, compositions and methods of treatment |
| US7098219B2 (en) | 2000-08-01 | 2006-08-29 | Wockhart Limited | Inhibitors of cellular efflux pumps of microbes |
| US6878713B2 (en) | 2001-04-25 | 2005-04-12 | Wockhardt Limited | Generation triple-targeting, chiral, broad-spectrum antimicrobial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation, compositions and use as medicaments |
| US7626032B2 (en) | 2001-04-25 | 2009-12-01 | Wockhardt Limited | Generation triple-targeting, chiral, broad-spectrum antimicrobial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation, compositions and use as medicaments |
| WO2002085886A3 (en) * | 2001-04-25 | 2003-05-22 | Souza Noel J De | Chiral, broad-spectrum antibacterial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation and compositions |
| WO2002085886A2 (en) * | 2001-04-25 | 2002-10-31 | Wockhardt Limited | Chiral, broad-spectrum antibacterial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation and compositions |
| US7393957B2 (en) | 2001-04-25 | 2008-07-01 | Wockhardt Limited | Generation triple-targeting, chiral, broad-spectrum antimicrobial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation, compositions and use as medicaments |
| US6964966B2 (en) | 2001-04-25 | 2005-11-15 | Wockhardt Limited | Generation triple-targeting, chiral, broad-spectrum antimicrobial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation, compositions and use as medicaments |
| WO2003050107A1 (en) * | 2001-12-13 | 2003-06-19 | Wockhardt Limited | New generation triple-targeting, chiral, broad-spectrum antimicrobial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation, compositions and use as medicaments |
| WO2003099815A1 (en) * | 2002-05-28 | 2003-12-04 | Wockhardt Limited | Crystalline fluoroquinolone arginine salt form |
| US6664267B1 (en) | 2002-05-28 | 2003-12-16 | Wockhardt Limited | Crystalline fluoroquinolone arginine salt form |
| EP1918289A3 (en) * | 2002-05-28 | 2009-08-19 | Wockhardt Limited | Crystalline nadifloxacin arginine salt form |
| US7132541B2 (en) | 2002-05-28 | 2006-11-07 | Wockhardt Limited | Crystalline fluoroquinolone arginine salt form |
| US7829543B2 (en) | 2003-01-07 | 2010-11-09 | Paratek Pharmaceuticals, Inc. | Substituted polyamines as inhibitors of bacterial efflux pumps |
| GB2403653A (en) * | 2003-07-10 | 2005-01-12 | Secr Defence | Use of fluoroquinone antibiotics for the treatment of plague |
| WO2005023805A1 (en) * | 2003-09-04 | 2005-03-17 | Wockhardt Limited | Benzoquinolizine-2-carboxylic acid arginine salt tetrahydrate |
| US7164023B2 (en) | 2003-09-04 | 2007-01-16 | Wockhardt Limited | Benzoquinolizine-2-carboxylic acid arginine salt tetrahydrate |
| HRP20060079B1 (en) * | 2003-09-04 | 2008-05-31 | Wockhardt Limited | Benzoquinolizine-2-carboxylic acid arginine salt tetrahydrate |
| WO2005026145A2 (en) * | 2003-09-12 | 2005-03-24 | Warner-Lambert Company Llc | Quinolone antibacterial agents |
| WO2005026145A3 (en) * | 2003-09-12 | 2005-06-16 | Warner Lambert Co | Quinolone antibacterial agents |
| WO2005070941A1 (en) * | 2004-01-13 | 2005-08-04 | Cumbre Pharmaceuticals Inc. | Rifamycin imino derivatives effective against drug-resistant microbes |
| US7238694B2 (en) | 2004-01-13 | 2007-07-03 | Cumbre Pharmaceuticals, Inc. | Rifamycin imino derivatives effective against drug-resistant microbes |
| US7247634B2 (en) | 2004-01-13 | 2007-07-24 | Cumbre Pharmaceuticals Inc. | Rifamycin derivatives effective against drug-resistant microbes |
| WO2005070940A3 (en) * | 2004-01-13 | 2005-09-29 | Cumbre Inc | Rifamycin derivatives effective against drug-resistant microbes |
| US7947741B2 (en) | 2004-03-17 | 2011-05-24 | Mpex Pharmaceuticals, Inc. | Use and administration of bacterial efflux pump inhibitors |
| WO2005089738A2 (en) * | 2004-03-17 | 2005-09-29 | Mpex Pharmaceuticals, Inc. | Use and administration of bacterial efflux pump inhibitors |
| US7994225B2 (en) | 2004-03-17 | 2011-08-09 | Rempex Pharmaceuticals, Inc. | Bacterial efflux pump inhibitors for the treatment of ophthalmic and otic infections |
| WO2005089738A3 (en) * | 2004-03-17 | 2007-08-23 | Mpex Pharmaceuticals Inc | Use and administration of bacterial efflux pump inhibitors |
| US7867992B2 (en) | 2004-07-21 | 2011-01-11 | Aicuris Gmbh & Co. Kg | Substituted quinolones |
| US7569563B2 (en) | 2005-06-30 | 2009-08-04 | Aicuris Gmbh & Co. Hk | Substituted quinolones II |
| US7977349B2 (en) | 2006-02-09 | 2011-07-12 | Aicuris Gmbh & Co. Kg | Substituted quinolones III |
| US9155792B2 (en) | 2006-02-13 | 2015-10-13 | Trustees Of Boston University | RecA inhibitors with antibiotic activity, compositions and methods of use |
| WO2007102061A3 (en) * | 2006-03-07 | 2009-04-23 | Wockhardt Ltd | Prodrugs of benzoquinolizine-2-carboxylic acid |
| US7868173B2 (en) | 2006-03-07 | 2011-01-11 | Wockhardt Limited | Prodrugs of benzoquinolizine-2-carboxylic acid |
| KR101364900B1 (en) * | 2006-03-07 | 2014-02-19 | 욱크하트 리미티드 | Prodrugs of benzoquinolizine-2-carboxylic acid |
| AU2007225088B2 (en) * | 2006-03-13 | 2012-09-13 | Kyorin Pharmaceutical Co., Ltd | Aminoquinolones as GSK-3 inhibitors |
| JP2009530292A (en) * | 2006-03-13 | 2009-08-27 | アクティヴィックス バイオサイエンシーズ インコーポレイテッド | Aminoquinolones as GSK-3 inhibitors |
| WO2007106537A3 (en) * | 2006-03-13 | 2007-11-29 | Activx Biosciences Inc | Aminoquinolones as gsk-3 inhibitors |
| US8063221B2 (en) | 2006-03-13 | 2011-11-22 | Kyorin Pharmaceutical Co., Ltd. | Aminoquinolones as GSK-3 inhibitors |
| WO2008019292A3 (en) * | 2006-08-04 | 2008-08-14 | Univ Boston | Compositions and methods for potentiating antibiotic activity |
| WO2008019292A2 (en) * | 2006-08-04 | 2008-02-14 | Trustees Of Boston University | Compositions and methods for potentiating antibiotic activity |
| US7902227B2 (en) | 2007-07-27 | 2011-03-08 | Janssen Pharmaceutica Nv. | C-7 isoxazolinyl quinolone / naphthyridine derivatives useful as antibacterial agents |
| US8389514B2 (en) | 2007-09-11 | 2013-03-05 | Kyorin Pharmaceutical Co., Ltd. | Cyanoaminoquinolones and tetrazoloaminoquinolones as GSK-3 inhibitors |
| CN102351880A (en) * | 2007-09-11 | 2012-02-15 | 杏林制药株式会社 | Cyanoaminoquinolones and tetrazoloaminoquinolones as GSK-3 inhibitors |
| CN102351880B (en) * | 2007-09-11 | 2014-11-12 | 杏林制药株式会社 | Cyanoaminoquinolones and tetrazoloaminoquinolones as GSK-3 inhibitors |
| US8901112B2 (en) | 2007-09-12 | 2014-12-02 | Kyorin Pharmaceutical Co., Ltd. | Spirocyclic aminoquinolones as GSK-3 inhibitors |
| US8476261B2 (en) | 2007-09-12 | 2013-07-02 | Kyorin Pharmaceutical Co., Ltd. | Spirocyclic aminoquinolones as GSK-3 inhibitors |
| US8071591B2 (en) | 2009-03-11 | 2011-12-06 | Kyorin Pharmaceutical Co., Ltd. | 7-cycloalkylaminoquinolones as GSK-3 inhibitors |
| WO2011101710A1 (en) * | 2010-02-16 | 2011-08-25 | Wockhardt Research Centre | Efflux pump inhibitors |
| EP2751083A4 (en) * | 2011-08-31 | 2015-06-10 | Otsuka Pharma Co Ltd | Quinolone compound |
| US9067887B2 (en) | 2011-08-31 | 2015-06-30 | Otsuka Pharmaceutical Co., Ltd | Quinolone compound |
| US9440951B2 (en) | 2011-08-31 | 2016-09-13 | Otsuka Pharmaceutical Co., Ltd | Quinolone compound |
| JP2017039733A (en) * | 2011-08-31 | 2017-02-23 | 大塚製薬株式会社 | Quinolone compound |
| CN103183684A (en) * | 2011-12-29 | 2013-07-03 | 杭州师范大学 | Quinolone lactone compound, and preparation method and application thereof |
| CN103183684B (en) * | 2011-12-29 | 2015-11-18 | 杭州师范大学 | A kind of quinlone lactone compound and its preparation method and application |
| CN109486739A (en) * | 2018-11-23 | 2019-03-19 | 上海海洋大学 | A method of induction vibrio parahemolyticus generates levofloxacin resistance |
| CN109486739B (en) * | 2018-11-23 | 2022-02-01 | 上海海洋大学 | Method for inducing vibrio parahaemolyticus to generate levofloxacin drug resistance |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1305048A2 (en) | 2003-05-02 |
| WO2002009758A3 (en) | 2002-12-27 |
| AU2001280091A1 (en) | 2002-02-13 |
| CA2417799A1 (en) | 2002-02-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2002009758A2 (en) | Inhibitors of cellular efflux pumps of microbes | |
| US7626032B2 (en) | Generation triple-targeting, chiral, broad-spectrum antimicrobial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation, compositions and use as medicaments | |
| US5137892A (en) | Quinoline, naphthyridine and pyridobenzoxazine derivatives | |
| ES2558685T3 (en) | Use of pyrroloquinoline compounds to eliminate clinically latent microorganisms | |
| US7247634B2 (en) | Rifamycin derivatives effective against drug-resistant microbes | |
| US7229996B2 (en) | Rifamycin derivatives | |
| US7820823B2 (en) | Dual action antibiotics | |
| KR101307717B1 (en) | Tri- or tetra-substituted-3-aminopyrrolidine derivatives | |
| US6964966B2 (en) | Generation triple-targeting, chiral, broad-spectrum antimicrobial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation, compositions and use as medicaments | |
| JP2012522807A (en) | Hydroxythienoquinolones and related compounds as antiinfectives | |
| NZ783605A (en) | Macrocyclic broad spectrum antibiotics | |
| JP2005511743A (en) | A new generation of triple-targeted chiral broad-spectrum antibacterial 7-substituted piperidino-quinolone carboxylic acid derivatives, methods for their preparation, compositions, and pharmaceutical use | |
| HU221952B1 (en) | 5-amino-8-methyl-7-pyrrolidinyl-quinoline-3-carboxylic acid derivatives and pharmaceutical compositions containing them | |
| US7098219B2 (en) | Inhibitors of cellular efflux pumps of microbes | |
| KR20180094042A (en) | Compound containing 7-oxo-6- (sulfooxy) -1,6-diazabicyclo [3.2.1] octane-2-carboxamide and its use for the treatment of bacterial infections | |
| RU2248970C2 (en) | Cycloalkyl-substituted derivatives of aminomethylpyrrolidine and antibacterial agent based on thereof | |
| EP0429304A2 (en) | Pyridone-carboxylic acid derivatives and their use as veterinary medicines | |
| US20120058989A1 (en) | Antibacterial fluoroquinolone analogs | |
| EP0302371B1 (en) | 7-(2-Methyl-4-aminopyrrolidinyl)naphthyridine and quinoline compounds | |
| JPH05132479A (en) | Amino acid azetidinyl substituted pyridone derivative, preparation thereof and medicinal application thereof | |
| Petersen | Quinolone antibiotics: the development of moxifloxacin | |
| Miyauchi et al. | Design, synthesis and biological evaluations of novel 7-[3-(1-aminocycloalkyl) pyrrolidin-1-yl]-6-desfluoro-8-methoxyquinolones with potent antibacterial activity against multi-drug resistant Gram-positive bacteria | |
| JP2021504489A (en) | Antibacterial heterocyclic compounds and their synthesis | |
| RU2029771C1 (en) | Optically active pyridobenzoxazine derivatives or salts thereof | |
| PT682030E (en) | 8-AMINO-10- (AZABICYLIC-ALKYL) -PYRIDY-1,2,3-D, E-1,3,4-BENZOXADIAZINE DERIVATIVES FOR THEIR PREPARATION AND ANTIBACTERIAL AGENTS CONTAINING THESE COMPOUNDS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2417799 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001958373 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001958373 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2001958373 Country of ref document: EP |