US8026347B2 - Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors - Google Patents
Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors Download PDFInfo
- Publication number
- US8026347B2 US8026347B2 US12/174,722 US17472208A US8026347B2 US 8026347 B2 US8026347 B2 US 8026347B2 US 17472208 A US17472208 A US 17472208A US 8026347 B2 US8026347 B2 US 8026347B2
- Authority
- US
- United States
- Prior art keywords
- formula
- compound
- optionally substituted
- substituted alkyl
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active, expires
Links
- 0 CC.CC.C[Y]C1=CC=CC=C1.[3*]CC1OC(C2=CC=CC=C2)[C@H](O)[C@@H](O)[C@@H]1O Chemical compound CC.CC.C[Y]C1=CC=CC=C1.[3*]CC1OC(C2=CC=CC=C2)[C@H](O)[C@@H](O)[C@@H]1O 0.000 description 18
- ZHDDJWFZDNKWIC-REIXXSIJSA-N CC1(C)O[C@@H]2O[C@@H](C(=O)N3CCOCC3)[C@@H](O)[C@@H]2O1 Chemical compound CC1(C)O[C@@H]2O[C@@H](C(=O)N3CCOCC3)[C@@H](O)[C@@H]2O1 ZHDDJWFZDNKWIC-REIXXSIJSA-N 0.000 description 3
- MFZMNZVLMNIVLH-UHFFFAOYSA-N CCOC1=CC=C(CC2=C(Cl)C=CC(C)=C2)C=C1 Chemical compound CCOC1=CC=C(CC2=C(Cl)C=CC(C)=C2)C=C1 MFZMNZVLMNIVLH-UHFFFAOYSA-N 0.000 description 3
- CZFRIPGHKLGMEU-UHFFFAOYSA-N CCOC1=CC=C(CC2=C(Cl)C=CC(I)=C2)C=C1 Chemical compound CCOC1=CC=C(CC2=C(Cl)C=CC(I)=C2)C=C1 CZFRIPGHKLGMEU-UHFFFAOYSA-N 0.000 description 3
- XVFWOGCQGKBZLV-UHFFFAOYSA-N CC.CC.CC1=CC=CC=C1.C[Y]C1=CC=CC=C1 Chemical compound CC.CC.CC1=CC=CC=C1.C[Y]C1=CC=CC=C1 XVFWOGCQGKBZLV-UHFFFAOYSA-N 0.000 description 2
- AQWPWGQLPICWFL-BHYPZZFFSA-N CC.CC.C[Y]C1=CC=CC=C1.POC1OC(C2=CC=CC=C2)[C@H](OP)[C@@H](OP)[C@@H]1OP Chemical compound CC.CC.C[Y]C1=CC=CC=C1.POC1OC(C2=CC=CC=C2)[C@H](OP)[C@@H](OP)[C@@H]1OP AQWPWGQLPICWFL-BHYPZZFFSA-N 0.000 description 2
- MSPDWQFAJOSSPO-OQDJIVLBSA-N CC.CC.C[Y]C1=CC=CC=C1.[H][C@@]1(C(=O)C2=CC=CC=C2)O[C@H]2OC(C)(C)O[C@H]2[C@@H]1O Chemical compound CC.CC.C[Y]C1=CC=CC=C1.[H][C@@]1(C(=O)C2=CC=CC=C2)O[C@H]2OC(C)(C)O[C@H]2[C@@H]1O MSPDWQFAJOSSPO-OQDJIVLBSA-N 0.000 description 2
- UCPZKXXFVQNJRP-HRFBMUIDSA-N CC1(C)O[C@@H]2O[C@@H](C(=O)N3CCOCC3)[C@@H](O)[C@@H]2O1.CC1(C)O[C@@H]2O[C@@H](C(=O)O)[C@@H](O)[C@@H]2O1.CC1(C)O[C@@H]2O[C@@H](CO)[C@@H](O)[C@@H]2O1.O=C[C@@H](O)[C@H](O)[C@@H](O)CO Chemical compound CC1(C)O[C@@H]2O[C@@H](C(=O)N3CCOCC3)[C@@H](O)[C@@H]2O1.CC1(C)O[C@@H]2O[C@@H](C(=O)O)[C@@H](O)[C@@H]2O1.CC1(C)O[C@@H]2O[C@@H](CO)[C@@H](O)[C@@H]2O1.O=C[C@@H](O)[C@H](O)[C@@H](O)CO UCPZKXXFVQNJRP-HRFBMUIDSA-N 0.000 description 2
- JAUQZVBVVJJRKM-VZFHVOOUSA-N CC1(C)O[C@@H]2O[C@@H](CO)[C@@H](O)[C@@H]2O1 Chemical compound CC1(C)O[C@@H]2O[C@@H](CO)[C@@H](O)[C@@H]2O1 JAUQZVBVVJJRKM-VZFHVOOUSA-N 0.000 description 2
- ABNIXBQXMYTWSJ-UIPNDDLNSA-N CCOC1=CC=C(CC2=CC([C@@H]3O[C@H](SC)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]3OC(C)=O)=CC=C2Cl)C=C1 Chemical compound CCOC1=CC=C(CC2=CC([C@@H]3O[C@H](SC)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]3OC(C)=O)=CC=C2Cl)C=C1 ABNIXBQXMYTWSJ-UIPNDDLNSA-N 0.000 description 2
- DKYUVICFVOYDEN-NQNKTMCVSA-N [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(CCC)[C@@H](O)[C@H](O)[C@H]1O Chemical compound [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(CCC)[C@@H](O)[C@H](O)[C@H]1O DKYUVICFVOYDEN-NQNKTMCVSA-N 0.000 description 2
- RTPYYCQAUNYHKG-QZQMKCFYSA-K B.Cl[Ce](Cl)Cl.O.O.O.O.O.O.O.[H][C@@]1(C(=O)C2=CC(CC3=CC=C(OCC)C=C3)=C(Cl)C=C2)C[C@H]2OC(C)(C)O[C@H]2[C@@H]1O.[H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(OC(C)=O)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O.[H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)O[C@H](SC)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O.[H][C@@]1([C@@H](O)C2=CC(CC3=CC=C(OCC)C=C3)=C(Cl)C=C2)O[C@H]2OC(C)(C)O[C@H]2[C@@H]1O.[NaH] Chemical compound B.Cl[Ce](Cl)Cl.O.O.O.O.O.O.O.[H][C@@]1(C(=O)C2=CC(CC3=CC=C(OCC)C=C3)=C(Cl)C=C2)C[C@H]2OC(C)(C)O[C@H]2[C@@H]1O.[H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(OC(C)=O)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O.[H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)O[C@H](SC)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O.[H][C@@]1([C@@H](O)C2=CC(CC3=CC=C(OCC)C=C3)=C(Cl)C=C2)O[C@H]2OC(C)(C)O[C@H]2[C@@H]1O.[NaH] RTPYYCQAUNYHKG-QZQMKCFYSA-K 0.000 description 1
- CJXOBPHRQGLXER-VOAKCMCISA-N CC(C)(O[C@H]1[C@@H]2O)O[C@@H]1O[C@H]2C(c1ccccc1)=O Chemical compound CC(C)(O[C@H]1[C@@H]2O)O[C@@H]1O[C@H]2C(c1ccccc1)=O CJXOBPHRQGLXER-VOAKCMCISA-N 0.000 description 1
- FRGCJBQQZRYAHU-WJYPOBIUSA-N CC.CC.C[Y]C1=CC=CC=C1.OC1OC(C2=CC=CC=C2)[C@H](O)[C@@H](O)[C@@H]1O Chemical compound CC.CC.C[Y]C1=CC=CC=C1.OC1OC(C2=CC=CC=C2)[C@H](O)[C@@H](O)[C@@H]1O FRGCJBQQZRYAHU-WJYPOBIUSA-N 0.000 description 1
- AEYZABZZCSJPSG-BHYPZZFFSA-N CC.CC.C[Y]C1=CC=CC=C1.PO[C@@H]1C(S)OC(C2=CC=CC=C2)[C@H](OP)[C@H]1OP Chemical compound CC.CC.C[Y]C1=CC=CC=C1.PO[C@@H]1C(S)OC(C2=CC=CC=C2)[C@H](OP)[C@H]1OP AEYZABZZCSJPSG-BHYPZZFFSA-N 0.000 description 1
- XYNGGWZMGMDNDO-FFKDCUGXSA-N CC.CC.C[Y]C1=CC=CC=C1.[H][C@@]1([C@@H](O)C2=CC=CC=C2)O[C@@H](C)[C@@H](OPP)[C@@H]1O Chemical compound CC.CC.C[Y]C1=CC=CC=C1.[H][C@@]1([C@@H](O)C2=CC=CC=C2)O[C@@H](C)[C@@H](OPP)[C@@H]1O XYNGGWZMGMDNDO-FFKDCUGXSA-N 0.000 description 1
- BRDPBVKINZOIJV-YLHTXXLFSA-N CC.CC.C[Y]C1=CC=CC=C1.[H][C@@]1([C@@H](O)C2=CC=CC=C2)O[C@@H](OPP)[C@@H](OPP)[C@@H]1O Chemical compound CC.CC.C[Y]C1=CC=CC=C1.[H][C@@]1([C@@H](O)C2=CC=CC=C2)O[C@@H](OPP)[C@@H](OPP)[C@@H]1O BRDPBVKINZOIJV-YLHTXXLFSA-N 0.000 description 1
- ORSDKHBXDQGCJU-PZTPTHGWSA-N CC.CC.C[Y]C1=CC=CC=C1.[H][C@@]1([C@@H](O)C2=CC=CC=C2)O[C@H]2OC(C)(C)O[C@H]2[C@@H]1O Chemical compound CC.CC.C[Y]C1=CC=CC=C1.[H][C@@]1([C@@H](O)C2=CC=CC=C2)O[C@H]2OC(C)(C)O[C@H]2[C@@H]1O ORSDKHBXDQGCJU-PZTPTHGWSA-N 0.000 description 1
- ZHDDJWFZDNKWIC-UHFFFAOYSA-N CC1(C)OC2OC(C(=O)N3CCOCC3)C(O)C2O1 Chemical compound CC1(C)OC2OC(C(=O)N3CCOCC3)C(O)C2O1 ZHDDJWFZDNKWIC-UHFFFAOYSA-N 0.000 description 1
- ODLRPZNUYNQMAC-RHDMBQCUSA-N CC1(C)O[C@@H]2O[C@@H](C(=O)N3CCOCC3)[C@@H](O)[C@@H]2O1.CCOC1=CC=C(CC2=C(Cl)C=CC(C(=O)[C@@H]3O[C@H]4OC(C)(C)O[C@H]4[C@@H]3O)=C2)C=C1.CCOC1=CC=C(CC2=C(Cl)C=CC(I)=C2)C=C1 Chemical compound CC1(C)O[C@@H]2O[C@@H](C(=O)N3CCOCC3)[C@@H](O)[C@@H]2O1.CCOC1=CC=C(CC2=C(Cl)C=CC(C(=O)[C@@H]3O[C@H]4OC(C)(C)O[C@H]4[C@@H]3O)=C2)C=C1.CCOC1=CC=C(CC2=C(Cl)C=CC(I)=C2)C=C1 ODLRPZNUYNQMAC-RHDMBQCUSA-N 0.000 description 1
- GEELQQUOYNRYCP-UHFFFAOYSA-N CC1=CC=C(CC2=C(Cl)C=CC(C)=C2)C=C1 Chemical compound CC1=CC=C(CC2=C(Cl)C=CC(C)=C2)C=C1 GEELQQUOYNRYCP-UHFFFAOYSA-N 0.000 description 1
- TUBVOYXRJBKZBN-UHFFFAOYSA-N CCOC1=CC=C(CC2=C(Cl)C=CC(C(=O)C3OC4OC(C)(C)OC4C3O)=C2)C=C1 Chemical compound CCOC1=CC=C(CC2=C(Cl)C=CC(C(=O)C3OC4OC(C)(C)OC4C3O)=C2)C=C1 TUBVOYXRJBKZBN-UHFFFAOYSA-N 0.000 description 1
- IGWZYKVVSUNCEM-UHFFFAOYSA-N CCOC1=CC=C(CC2=C(Cl)C=CC(C(O)C3OC4OC(C)(C)OC4C3O)=C2)C=C1 Chemical compound CCOC1=CC=C(CC2=C(Cl)C=CC(C(O)C3OC4OC(C)(C)OC4C3O)=C2)C=C1 IGWZYKVVSUNCEM-UHFFFAOYSA-N 0.000 description 1
- DEYSVLVWOGCWDN-UHFFFAOYSA-M CCOC1=CC=C(CC2=C(Cl)C=CC([Mg]Cl)=C2)C=C1 Chemical compound CCOC1=CC=C(CC2=C(Cl)C=CC([Mg]Cl)=C2)C=C1 DEYSVLVWOGCWDN-UHFFFAOYSA-M 0.000 description 1
- SLGJUMBXNGOSHR-UHFFFAOYSA-N CCOC1=CC=C(CC2=CC(C3OC(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C3OC(C)=O)=CC=C2Cl)C=C1 Chemical compound CCOC1=CC=C(CC2=CC(C3OC(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C3OC(C)=O)=CC=C2Cl)C=C1 SLGJUMBXNGOSHR-UHFFFAOYSA-N 0.000 description 1
- WJZRFTOJWJBWEX-UHFFFAOYSA-N CCOC1=CC=C(CC2=CC(C3OC(S)C(OC(C)=O)C(OC(C)=O)C3OC(C)=O)=CC=C2Cl)C=C1 Chemical compound CCOC1=CC=C(CC2=CC(C3OC(S)C(OC(C)=O)C(OC(C)=O)C3OC(C)=O)=CC=C2Cl)C=C1 WJZRFTOJWJBWEX-UHFFFAOYSA-N 0.000 description 1
- WUNDKHVIULDKQM-UHFFFAOYSA-N CCOC1=CC=C(CC2=CC(C3OC(SC)C(C)C(OC(C)=O)C3OC(C)=O)=CC=C2Cl)C=C1 Chemical compound CCOC1=CC=C(CC2=CC(C3OC(SC)C(C)C(OC(C)=O)C3OC(C)=O)=CC=C2Cl)C=C1 WUNDKHVIULDKQM-UHFFFAOYSA-N 0.000 description 1
- LITRERKYGZPHDC-CRSSMBPESA-N CCOc1ccc(Cc2cc([C@@H]([C@@H]([C@H]([C@@H]3O)O)O)O[C@@H]3S(C)(=O)=O)ccc2Cl)cc1 Chemical compound CCOc1ccc(Cc2cc([C@@H]([C@@H]([C@H]([C@@H]3O)O)O)O[C@@H]3S(C)(=O)=O)ccc2Cl)cc1 LITRERKYGZPHDC-CRSSMBPESA-N 0.000 description 1
- TUBVOYXRJBKZBN-MLNNCEHLSA-N [H][C@@]1(C(=O)C2=CC(CC3=CC=C(OCC)C=C3)=C(Cl)C=C2)O[C@H]2OC(C)(C)O[C@H]2[C@@H]1O Chemical compound [H][C@@]1(C(=O)C2=CC(CC3=CC=C(OCC)C=C3)=C(Cl)C=C2)O[C@H]2OC(C)(C)O[C@H]2[C@@H]1O TUBVOYXRJBKZBN-MLNNCEHLSA-N 0.000 description 1
- ALXLZBYULBUCMB-VANKVMQKSA-N [H][C@@]1(C(C)=O)O[C@@H](OPP)[C@@H](OPP)[C@@H]1O Chemical compound [H][C@@]1(C(C)=O)O[C@@H](OPP)[C@@H](OPP)[C@@H]1O ALXLZBYULBUCMB-VANKVMQKSA-N 0.000 description 1
- PBKNTYIKAAJDSY-ONHKFNRTSA-N [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(C)[C@@H](C)[C@H](OC(C)=O)[C@H]1OC(C)=O Chemical compound [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(C)[C@@H](C)[C@H](OC(C)=O)[C@H]1OC(C)=O PBKNTYIKAAJDSY-ONHKFNRTSA-N 0.000 description 1
- YCEUTZKYUQXUCF-OETYQTQASA-N [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(C)[C@@H](O)[C@H](O)[C@H]1O Chemical compound [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(C)[C@@H](O)[C@H](O)[C@H]1O YCEUTZKYUQXUCF-OETYQTQASA-N 0.000 description 1
- WQMNRKXASOHNAU-IZGWVBKVSA-N [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(C)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O Chemical compound [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(C)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O WQMNRKXASOHNAU-IZGWVBKVSA-N 0.000 description 1
- BMCKPGKPUZNIBS-DSBNJHONSA-N [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(CC)[C@@H](O)[C@H](O)[C@H]1O Chemical compound [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(CC)[C@@H](O)[C@H](O)[C@H]1O BMCKPGKPUZNIBS-DSBNJHONSA-N 0.000 description 1
- FYQNAPUGYQIAMQ-OBPGIKJGSA-N [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(OC(C)=O)[C@@H](C)[C@H](OC(C)=O)[C@H]1OC(C)=O Chemical compound [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(OC(C)=O)[C@@H](C)[C@H](OC(C)=O)[C@H]1OC(C)=O FYQNAPUGYQIAMQ-OBPGIKJGSA-N 0.000 description 1
- SLGJUMBXNGOSHR-DOEHWDRQSA-N [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(OC(C)=O)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O Chemical compound [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)OC(OC(C)=O)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O SLGJUMBXNGOSHR-DOEHWDRQSA-N 0.000 description 1
- NYLIVNVMDJVTFW-NRSLGRGUSA-N [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)O[C@H](S(C)(=O)=O)[C@@H](O)[C@H](O)[C@H]1O.[H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)O[C@H](SC)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O Chemical compound [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)O[C@H](S(C)(=O)=O)[C@@H](O)[C@H](O)[C@H]1O.[H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)O[C@H](SC)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O NYLIVNVMDJVTFW-NRSLGRGUSA-N 0.000 description 1
- DKAKXBDXWIRTLU-NRSLGRGUSA-N [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)O[C@H](SC)[C@@H](O)[C@H](O)[C@H]1O.[H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)O[C@H](SC)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O Chemical compound [H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)O[C@H](SC)[C@@H](O)[C@H](O)[C@H]1O.[H][C@@]1(C2=CC=C(Cl)C(CC3=CC=C(OCC)C=C3)=C2)O[C@H](SC)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O DKAKXBDXWIRTLU-NRSLGRGUSA-N 0.000 description 1
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H5/00—Compounds containing saccharide radicals in which the hetero bonds to oxygen have been replaced by the same number of hetero bonds to halogen, nitrogen, sulfur, selenium, or tellurium
- C07H5/04—Compounds containing saccharide radicals in which the hetero bonds to oxygen have been replaced by the same number of hetero bonds to halogen, nitrogen, sulfur, selenium, or tellurium to nitrogen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/02—Acyclic radicals, not substituted by cyclic structures
- C07H15/04—Acyclic radicals, not substituted by cyclic structures attached to an oxygen atom of the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/04—Carbocyclic radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H9/00—Compounds containing a hetero ring sharing at least two hetero atoms with a saccharide radical
- C07H9/02—Compounds containing a hetero ring sharing at least two hetero atoms with a saccharide radical the hetero ring containing only oxygen as ring hetero atoms
- C07H9/04—Cyclic acetals
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- This invention relates to methods of preparing inhibitors of sodium glucose co-transporter 2.
- the sodium glucose co-transporter 2 (SGLT2) is a transporter that reabsorbs glucose from the renal filtrate and prevents the loss of glucose in the urine. Because competitive inhibitors of SGLT2 cause the renal excretion of glucose, they may be used to normalize high blood glucose levels associated with diseases such as diabetes. Handlon, A. L., Expert Opin. Ther. Patents 15(11):1531-1540 (2005).
- SGLT2 inhibitors In the search for new drugs that may be used to treat diabetes, a number of SGLT2 inhibitors have been disclosed. See, e.g., Handlon, supra; U.S. Pat. No. 6,515,117; U.S. patent application publication nos. US 2006/0035841, US 2004/0138439. At least one inhibitor is in clinical development as a treatment for Type 2 diabetes mellitus.
- This invention encompasses methods of preparing certain inhibitors of SGLT2, as well as compounds useful therein.
- FIG. 1 is a X-ray diffraction pattern of a crystalline solid form of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate.
- the spectrum was obtained using a Rigaku MiniFlex diffractometer (Cu (1.54060 ⁇ ) radiation).
- FIG. 2 is a X-ray diffraction pattern of a crystalline solid form of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone.
- the spectrum was obtained using a Rigaku MiniFlex diffractometer (Cu (1.54060 ⁇ ) radiation).
- FIG. 3 is a X-ray diffraction pattern of a crystalline solid form of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone.
- the spectrum was obtained using a Rigaku MiniFlex diffractometer (Cu (1.54060 ⁇ ) radiation).
- FIG. 4 is a X-ray diffraction pattern of a crystalline solid form of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene. The spectrum was obtained using a Rigaku MiniFlex diffractometer (Cu (1.54060 ⁇ ) radiation).
- Novel compounds that inhibit the sodium glucose co-transporter 2 were recently disclosed. See U.S. provisional application Nos. 60/848,156, filed Sep. 29, 2006, and 60/905,714, filed Mar. 8, 2007. This invention is based, in part, on the discovery of new methods of preparing those compounds. Particular methods of the invention allow for the compounds' large-scale manufacture.
- alkenyl means a straight chain, branched and/or cyclic hydrocarbon having from 2 to 20 (e.g., 2 to 10 or 2 to 6) carbon atoms, and including at least one carbon-carbon double bond.
- alkenyl moieties include vinyl, allyl, 1-butenyl, 2-butenyl, isobutylenyl, 1-pentenyl, 2-pentenyl, 3-methyl-1-butenyl, 2-methyl-2-butenyl, 2,3-dimethyl-2-butenyl, 1-hexenyl, 2-hexenyl, 3-hexenyl, 1-heptenyl, 2-heptenyl, 3-heptenyl, 1-octenyl, 2-octenyl, 3-octenyl, 1-nonenyl, 2-nonenyl, 3-nonenyl, 1-decenyl, 2-decenyl and 3-decenyl.
- alkoxy means an —O-alkyl group.
- alkoxy groups include, but are not limited to, —OCH 3 , —OCH 2 CH 3 , —O(CH 2 ) 2 CH 3 , O(CH 2 ) 3 CH 3 , —O(CH 2 ) 4 CH 3 , and —O(CH 2 ) 5 CH 3 .
- alkyl means a straight chain, branched and/or cyclic (“cycloalkyl”) hydrocarbon having from 1 to 20 (e.g., 1 to 10 or 1 to 4) carbon atoms. Alkyl moieties having from 1 to 4 carbons are referred to as “lower alkyl.” Examples of alkyl groups include, but are not limited to, methyl, ethyl, propyl, isopropyl, n-butyl, t-butyl, isobutyl, pentyl, hexyl, isohexyl, heptyl, 4,4-dimethylpentyl, octyl, 2,2,4-trimethylpentyl, nonyl, decyl, undecyl and dodecyl.
- Cycloalkyl moieties may be monocyclic or multicyclic, and examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and adamantyl. Additional examples of alkyl moieties have linear, branched and/or cyclic portions (e.g., 1-ethyl-4-methyl-cyclohexyl).
- alkyl includes saturated hydrocarbons as well as alkenyl and alkynyl moieties.
- alkylaryl or “alkyl-aryl” means an alkyl moiety bound to an aryl moiety.
- alkylheteroaryl or “alkyl-heteroaryl” means an alkyl moiety bound to a heteroaryl moiety.
- alkylheterocycle or “alkyl-heterocycle” means an alkyl moiety bound to a heterocycle moiety.
- alkynyl means a straight chain, branched or cyclic hydrocarbon having from 2 to 20 (e.g., 2 to 20 or 2 to 6) carbon atoms, and including at least one carbon-carbon triple bond.
- alkynyl moieties include acetylenyl, propynyl, 1-butynyl, 2-butynyl, 1-pentynyl, 2-pentynyl, 3-methyl-1-butynyl, 4-pentynyl, 1-hexynyl, 2-hexynyl, 5-hexynyl, 1-heptynyl, 2-heptynyl, 6-heptynyl, 1-octynyl, 2-octynyl, 7-octynyl, 1-nonynyl, 2-nonynyl, 8-nonynyl, 1-decynyl, 2-decynyl and 9-decynyl.
- aryl means an aromatic ring or an aromatic or partially aromatic ring system composed of carbon and hydrogen atoms.
- An aryl moiety may comprise multiple rings bound or fused together.
- aryl moieties include, but are not limited to, anthracenyl, azulenyl, biphenyl, fluorenyl, indan, indenyl, naphthyl, phenanthrenyl, phenyl, 1,2,3,4-tetrahydro-naphthalene, and tolyl.
- arylalkyl or “aryl-alkyl” means an aryl moiety bound to an alkyl moiety.
- halogen and “halo” encompass fluorine, chlorine, bromine, and iodine.
- heteroalkyl refers to an alkyl moiety (e.g., linear, branched or cyclic) in which at least one of its carbon atoms has been replaced with a heteroatom (e.g., N, O or S).
- heteroaryl means an aryl moiety wherein at least one of its carbon atoms has been replaced with a heteroatom (e.g., N, O or S).
- heteroatom e.g., N, O or S.
- examples include, but are not limited to, acridinyl, benzimidazolyl, benzofuranyl, benzoisothiazolyl, benzoisoxazolyl, benzoquinazolinyl, benzothiazolyl, benzoxazolyl, furyl, imidazolyl, indolyl, isothiazolyl, isoxazolyl, oxadiazolyl, oxazolyl, phthalazinyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrimidyl, pyrrolyl, quinazolinyl, quinolinyl, tetrazolyl,
- heteroarylalkyl or “heteroaryl-alkyl” means a heteroaryl moiety bound to an alkyl moiety.
- heterocycle refers to an aromatic, partially aromatic or non-aromatic monocyclic or polycyclic ring or ring system comprised of carbon, hydrogen and at least one heteroatom (e.g., N, O or S).
- a heterocycle may comprise multiple (i.e., two or more) rings fused or bound together.
- Heterocycles include heteroaryls.
- Examples include, but are not limited to, benzo[1,3]dioxolyl, 2,3-dihydro-benzo[1,4]dioxinyl, cinnolinyl, furanyl, hydantoinyl, morpholinyl, oxetanyl, oxiranyl, piperazinyl, piperidinyl, pyrrolidinonyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, tetrahydropyridinyl, tetrahydropyrimidinyl, tetrahydrothiophenyl, tetrahydrothiopyranyl and valerolactamyl.
- heterocyclealkyl or “heterocycle-alkyl” refers to a heterocycle moiety bound to an alkyl moiety.
- heterocycloalkyl refers to a non-aromatic heterocycle.
- heterocycloalkylalkyl or “heterocycloalkyl-alkyl” refers to a heterocycloalkyl moiety bound to an alkyl moiety.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic acids or bases including inorganic acids and bases and organic acids and bases.
- suitable pharmaceutically acceptable base addition salts include, but are not limited to, metallic salts made from aluminum, calcium, lithium, magnesium, potassium, sodium and zinc or organic salts made from lysine, N,N′-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, meglumine (N-methylglucamine) and procaine.
- Suitable non-toxic acids include, but are not limited to, inorganic and organic acids such as acetic, alginic, anthranilic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethenesulfonic, formic, fumaric, furoic, galacturonic, gluconic, glucuronic, glutamic, glycolic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phenylacetic, phosphoric, propionic, salicylic, stearic, succinic, sulfanilic, sulfuric, tartaric acid, and p-toluenesulfonic acid.
- inorganic and organic acids such as acetic, alginic, anthranilic, benzenesulfonic, benzoic, camphorsulfonic
- Non-toxic acids include hydrochloric, hydrobromic, phosphoric, sulfuric, and methanesulfonic acids.
- Examples of specific salts thus include hydrochloride and mesylate salts.
- Others are well-known in the art. See, e.g., Remington's Pharmaceutical Sciences, 18 th ed. (Mack Publishing, Easton Pa.: 1990) and Remington: The Science and Practice of Pharmacy, 19 th ed. (Mack Publishing, Easton Pa.: 1995).
- stereomerically pure means a composition that comprises one stereoisomer of a compound and is substantially free of other stereoisomers of that compound.
- a stereomerically pure composition of a compound having one stereocenter will be substantially free of the opposite stereoisomer of the compound.
- a stereomerically pure composition of a compound having two stereocenters will be substantially free of other diastereomers of the compound.
- a typical stereomerically pure compound comprises greater than about 80% by weight of one stereoisomer of the compound and less than about 20% by weight of other stereoisomers of the compound, greater than about 90% by weight of one stereoisomer of the compound and less than about 10% by weight of the other stereoisomers of the compound, greater than about 95% by weight of one stereoisomer of the compound and less than about 5% by weight of the other stereoisomers of the compound, greater than about 97% by weight of one stereoisomer of the compound and less than about 3% by weight of the other stereoisomers of the compound, or greater than about 99% by weight of one stereoisomer of the compound and less than about 1% by weight of the other stereoisomers of the compound.
- substituted when used to describe a chemical structure or moiety, refers to a derivative of that structure or moiety wherein one or more of its hydrogen atoms is substituted with a chemical moiety or functional group such as, but not limited to, alcohol, aldehyde, alkoxy, alkanoyloxy, alkoxycarbonyl, alkenyl, alkyl (e.g., methyl, ethyl, propyl, t-butyl), alkynyl, alkylcarbonyloxy (—OC(O)alkyl), amide (—C(O)NH-alkyl- or -alkylNHC(O)alkyl), amidinyl (—C(NH)NH-alkyl- or —C(NR)NH 2 ), amine (primary, secondary and tertiary such as alkylamino, arylamino, arylalkylamino), aroyl, aryl, ary
- the term “include” has the same meaning as “include, but are not limited to,” and the term “includes” has the same meaning as “includes, but is not limited to.” Similarly, the term “such as” has the same meaning as the term “such as, but not limited to.”
- one or more adjectives immediately preceding a series of nouns is to be construed as applying to each of the nouns.
- the phrase “optionally substituted alky, aryl, or heteroaryl” has the same meaning as “optionally substituted alky, optionally substituted aryl, or optionally substituted heteroaryl.”
- a chemical moiety that forms part of a larger compound may be described herein using a name commonly accorded it when it exists as a single molecule or a name commonly accorded its radical.
- the terms “pyridine” and “pyridyl” are accorded the same meaning when used to describe a moiety attached to other chemical moieties.
- the two phrases “XOH, wherein X is pyridyl” and “XOH, wherein X is pyridine” are accorded the same meaning, and encompass the compounds pyridin-2-ol, pyridin-3-ol and pyridin-4-ol.
- any atom shown in a drawing with unsatisfied valences is assumed to be attached to enough hydrogen atoms to satisfy the valences.
- chemical bonds depicted with one solid line parallel to one dashed line encompass both single and double (e.g., aromatic) bonds, if valences permit.
- This invention encompasses methods of preparing compounds of formula I:
- Y is O, S, NR 4 , or C(R 4 ) 2 ;
- Z 1 is O, S, SO, or SO 2 ;
- each R 1 is independently hydrogen, halogen, cyano, OR 1A , SR 1A , or optionally substituted alkyl;
- each R 1A is independently hydrogen or optionally substituted alkyl or aryl;
- each R 2 is independently hydrogen, halogen, cyano, OR 2A , SR 2A , or optionally substituted alkyl;
- each R 2A is independently hydrogen or optionally substituted alkyl or aryl;
- R 3 is optionally substituted alkyl, aryl or heterocycle;
- each R 4 is independently hydrogen or optionally substituted alkyl or aryl;
- each P 1 is independently a hydroxyl protecting group stable under acidic conditions.
- a compound of formula II(a) is oxidized to provide a compound of formula II, which is then contacted with a base to afford the compound of formula I.
- Suitable oxidation conditions are known in the art, and include the use of peroxy compounds, such as m-chlorophenylperacid, peracetic acid, oxone, a mixture of hydrogen peroxide or its complex (e.g., urea hydrogen peroxide) and acid anhydride (e.g., phthalic anhydride).
- Suitable bases are also known in the art, and include alkoxides, hydroxide, carbonates and amines.
- moieties encompassed by the definitions of various moieties may be protected using methods known in the art.
- the final product may undergo further reactions known in the art to afford other compounds encompassed by formula I.
- the final product may also be crystallized. In one method, the product is co-crystallized with an amino acid (e.g., L-phenylalanine, L-phenylglycine, L-arginine).
- an amino acid e.g., L-phenylalanine, L-phenylglycine, L-arginine
- Y is C(R 4 ) 2 .
- Z 1 is S, SO or SO 2 .
- each P 1 is independently C(O)R 5 , wherein each R 5 is independently alkyl, aryl, alkylaryl, or arylalkyl. Examples of P 1 include acetyl, benzoyl and pivaloyl.
- R 1 is OR 1A and R 1A is, for example, optionally substituted lower alkyl.
- R 2 is halogen.
- R 3 is lower alkyl (e.g., methyl or ethyl).
- R 4 is hydrogen.
- m is 1.
- n is 1.
- Y is CH 2
- Z 1 is S or SO 2
- R 1 is ethoxy
- R 2 is chloro
- R 3 is methyl
- Y is CH 2
- Z 1 is S or SO 2
- R 1 is ethoxy
- R 2 is chloro
- R 3 is ethyl
- the compound of formula II(a) is of formula II(b), which can be prepared as shown below in Scheme 2:
- reaction conditions are known in the art.
- basic conditions e.g., the use of a base such as N,N-diisopropyl ethylamine
- the compound of formula II(b) is of the formula:
- a specific compound of formula II(b) is (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate:
- a particular crystalline form of this compound has a melting point of about 156° C. as measured by differential scanning calorimetry (DSC) (onset temperature).
- the form provides a X-ray powder diffraction (XRPD) pattern with peaks at one or more of about 7.7, 11.9, 12.4, 16.9, 19.5, 19.9, 21.9, 23.2, 24.1, and/or 27.7 degrees 2 ⁇ .
- XRPD X-ray powder diffraction
- the relative intensities of peaks in a X-ray diffraction pattern of a crystalline form can vary depending on how the sample is prepared and how the data is collected. With this in mind, an example of a XRPD pattern of this crystalline form is provided in FIG. 1 .
- each P 2 is independently a hydroxyl protecting group stable under acidic conditions, or both P 2 s are taken together to provide a single protecting group;
- X′ is chlorine, bromine or iodine; and
- X′′ is a leaving group (e.g., amino, alkoxyamino, hydroxy, halogen, alkoxy, phenoxy, carboxy, sulfoxy).
- each P 2 is independently C(O)R 6 , or both P 2 s are taken together to provide C(R 6 ) 2 , wherein each R 6 is independently alkyl, aryl, alkylaryl, or arylalkyl.
- the compound of formula II(a) is obtained by contacting a compound of formula II(d) with reagents and reaction conditions that will depend on the nature of Z 2 .
- the compound of formula II(d) can be contacted with a Lewis acid (e.g., trimethylsilyl trifluoromethanesulfonate) and thiourea to obtain a compound wherein Z 2 is S (e.g., of formula II(c), shown above in Scheme 2).
- the compound of formula II(d) can be contacted with a hydroxyl compound under acidic conditions to afford a compound wherein Z 2 is O.
- Compounds of formula II(d) can be obtained by contacting a compound of formula II(e) with P 1 —X′′′ under suitable reaction conditions, wherein X′′′ is chlorine, bromine, iodine, alkylcarboxy, alkanesulfoxy, or alkoxysulfoxy.
- suitable reaction conditions are known in the art.
- the reaction may be catalyzed by a base, such as pyridine.
- the compound of formula II(d) is of the formula:
- the salt of P 1 is, for example, acyl chloride or acetic anhydride.
- Compounds of formula II(e) can be prepared by contacting a compound of formula III(a) with an acid under conditions sufficient to provide the compound of formula II(e).
- Suitable acids are known in the art, and include acetic acid, hydrochloric acid, sulfuric acid, phosphoric acid, methanesulfonic acid, and toluenesulfonic acid.
- Compounds of formula III(a) can be prepared by reducing a compound of formula III(b). Suitable reducing conditions are known in the art, and include the use of cerium chloride and sodium borohydride, a borane complex, enzymatic reduction, and hydrogenation or transfer hydrogenation.
- the compound of formula III(b) is of the formula:
- a specific compound of formula III(b) is (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone:
- a particular crystalline form of this compound has a melting point of about 113° C. as measured by DSC (onset temperature).
- the form provides a XRPD pattern with peaks at one or more of about 7.6, 13.2, 17.0, 17.4, 18.6, 19.5, 20.5, 20.8 and/or 23.2 degrees 2 ⁇ .
- An example of a XRPD pattern of this crystalline form is provided in FIG. 2 .
- Compounds of formula III(b) can be prepared by coupling a compound of formula IV with a compound of formula V.
- Suitable coupling conditions are known in the art, and include the use of metalating (e.g., magnesium or lithium) or transmetalating agents such as magnesium reagents (e.g., alkyl magnesium halide, dialkyl magnesium, lithium trialkyl magnesium halide) and organolithium reagents (e.g., n-butyl lithium, sec-butyl lithium, t-butyl lithium).
- metalating e.g., magnesium or lithium
- transmetalating agents such as magnesium reagents (e.g., alkyl magnesium halide, dialkyl magnesium, lithium trialkyl magnesium halide) and organolithium reagents (e.g., n-butyl lithium, sec-butyl lithium, t-butyl lithium).
- organolithium reagents e.g., n-butyl lithium
- M is an appropriate metal, such as Na, K, Li, or Mg
- X′ is Cl, Br, or I
- p is 0, 1, or 2, depending on the metal.
- the compound of formula V is such that X′′ is amino (e.g., morpholino).
- a specific compound of formula V is ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone:
- a particular crystalline form of this compound has a melting point of about 136° C. as measured by DSC (onset temperature).
- the form provides a XRPD pattern with peaks at one or more of about 9.0, 16.9, 17.6, 18.2, 18.4, 18.8 and/or 22.7 degrees 2 ⁇ .
- An example of a XRPD pattern of this crystalline form is provided in FIG. 3 .
- Suitable reaction conditions are known in the art, and include those described below in the Examples.
- L-( ⁇ )-xylose is cyclized under conditions sufficient to provide compound 1, which is then oxidized to provide compound 2, which is then contacted with morpholine under conditions sufficient to provide ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone.
- This invention encompasses compounds of formulae 1 and 2, including crystalline forms thereof.
- a specific compound of formula IV is 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene:
- a particular crystalline form of this compound has a melting point of about 65° C. (as determined by m.p. apparatus).
- the form provides a XRPD pattern with peaks at one or more of about 5.1, 13.5, 15.2, 20.3, 22.2 and/or 27.0 degrees 2 ⁇ .
- An example of a XRPD pattern of this crystalline form is provided in FIG. 4 .
- Particular compounds of formula IV(a) include those of the formula:
- the yellow oil was suspended in 2.5 L water stirring in a 5 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler.
- the pH was adjusted from 9 to 2 with 1N aq. HCl (142 mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5 S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol.
- a solution of NaClO 2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K 2 HPO 4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling.
- Bleach (3.0 L, approximate 6% household grade) was mixed with the K 2 HPO 4 solution.
- the addition rate of the NaClO 2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K 2 HPO 4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25° C. Additional charges of TEMPO (14.3 g, 0.5 mol %) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to ⁇ 10° C. and quenched with Na 2 SO 3 (1.4 kg, 0.6 eq) in three portions over 1 hr.
- the reaction mixture was then acidified with H 3 PO 4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15° C.
- the layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L ⁇ 3).
- the combined organic layer was concentrated under vacuo ( ⁇ 100-120 torr) at ⁇ 35° C. (28-32° C. vapor, 45-50° C. bath) to low volume ( ⁇ 6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached ⁇ 1% when diluted to volume of about 12-15 L with acetonitrile.
- Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen.
- the reaction mixture was added to 10 wt % aqueous NH 4 Cl (10 L, 5 ⁇ ) at 0° C. with vigorous stirring, and stirred for 30 minutes at 0° C.
- To this mixture was added slowly 6 N HCl (4 L, 2 ⁇ ) at 0° C. to obtain a clear solution and stirred for 30 minutes at 10° C.
- the organic layer was washed with 25 wt % aq NaCl (5 L, 2.5 ⁇ ). Then the organic layer was concentrated to a 3 ⁇ solution under the conditions (200 mbar, bath temp 50° C.).
- EtOAc 24 L, 12 ⁇ was added, and evaporated to a 3 ⁇ solution under the conditions (150 mbar, bath temp 50° C.).
- EtOAc (4 L, 2 ⁇ ) was added and concentrated to dryness (150 mbar, bath temp 50° C.). The wet cake was then transferred to a 50 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet. After EtOAc was added, the suspension was heated at 70° C. to obtain a 2.5 ⁇ homogeneous solution. To the resulting homogeneous solution was added slowly heptane (5 L, 2.5 ⁇ ) at the same temperature. A homogeneous solution was seeded and heptane (15 L, 7.5 ⁇ ) was added slowly to a little cloudy solution at 70° C. After stirred for 0.5 h at 70° C., the suspension was slowly cooled to 60° C.
- TMSOTf trimethylsilyl trifluoromethanesulfonate
- the reaction was quenched with 10 wt % aq NH 4 Cl (2.5 L, 1 ⁇ ) and the mixture was concentrated under vacuum to 5 ⁇ , diluted with water (10 L, 4 ⁇ ) and MTBE (12.5 L, 5 ⁇ ). The mixture was cooled to 10° C. and 6 N aq HCl was added until the pH of the mixture reached 2.0. Stirring was continued for 10 minutes and the layers were separated. The organic layer was washed with H 2 O (5 L, 2 ⁇ ). The combined aqueous layer was extracted with MTBE (12.5 L, 5 ⁇ ). The combined organic layers were washed with brine (2.5 L, 1 ⁇ ) and concentrated under vacuum to 3 ⁇ . MeCN (15 L, 6 ⁇ ) was added. The mixture was concentrated again to 10 L (4 ⁇ ) and any solid residue was removed by a polish filtration. The cake was washed with minimal amount of MeCN.
- the organic filtrate was transferred to 50 L reactor, and a pre-prepared 20 mol % aqueous H 2 SO 4 solution (61.8 mL 98% concentrated H 2 SO 4 and 5 L H 2 O) was added.
- the mixture was heated to 80° C. for 2 hours and then cooled to 20° C.
- the reaction was quenched with a solution of saturated aqueous K 2 CO 3 (5 L, 2 ⁇ ) and diluted with MTBE (15 L, 6 ⁇ ).
- the organic layer was separated, washed with brine (5 L, 2 ⁇ ) and concentrated under vacuum to 5 L (2 ⁇ ). MeCN (12.5 L, 5 ⁇ ) was added and the mixture was concentrated to 7.5 L (3 ⁇ ).
- MEK (100 kg) was added and the same about of solvent was distilled under vacuum. This MEK addition and distillation was repeated to dry the solution. Enough MEK was added to produce a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol in 50 L MEK. This solution was polish filtered and heptane (100 L) was added at about 80° C. Form 2 seeds (0.1 kg) were added followed by slow addition of heptane (100 L) as 80° C. Heating was continued for 8 h more at 80° C., cooled to 20° C. over at least 3 hours, held at this temperature for at least 2 hours, filtered, and washed with MEK/heptane. The cake was dried at 50° C. under vacuum to afford the title compound as a white solid (6.6 kg, 86% yield).
Abstract

Description

the substitutes of which are defined herein, and salts thereof, which comprises contacting a compound of formula II:

with a base under suitable conditions.

and salts and co-crystals thereof, wherein: Y is O, S, NR4, or C(R4)2; Z1 is O, S, SO, or SO2; each R1 is independently hydrogen, halogen, cyano, OR1A, SR1A, or optionally substituted alkyl; each R1A is independently hydrogen or optionally substituted alkyl or aryl; each R2 is independently hydrogen, halogen, cyano, OR2A, SR2A, or optionally substituted alkyl; each R2A is independently hydrogen or optionally substituted alkyl or aryl; R3 is optionally substituted alkyl, aryl or heterocycle; each R4 is independently hydrogen or optionally substituted alkyl or aryl; n is 1-3; and m is 1-3.

wherein each P1 is independently a hydroxyl protecting group stable under acidic conditions. In this approach, a compound of formula II(a) is oxidized to provide a compound of formula II, which is then contacted with a base to afford the compound of formula I. Suitable oxidation conditions are known in the art, and include the use of peroxy compounds, such as m-chlorophenylperacid, peracetic acid, oxone, a mixture of hydrogen peroxide or its complex (e.g., urea hydrogen peroxide) and acid anhydride (e.g., phthalic anhydride). Suitable bases are also known in the art, and include alkoxides, hydroxide, carbonates and amines.
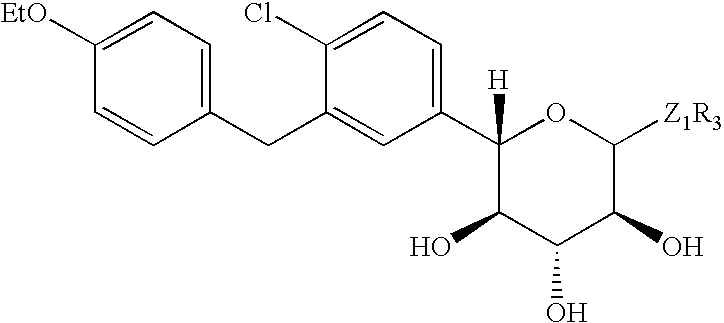
In another embodiment, Y is CH2, Z1 is S or SO2, R1 is ethoxy, R2 is chloro, and R3 is ethyl. For example, in a particular method, the compound of formula I is of the formula:


wherein X is bromine, iodine, alkanesulfoxy, or alkoxysulfoxy. Suitable reaction conditions are known in the art. For example, basic conditions (e.g., the use of a base such as N,N-diisopropyl ethylamine) can be used. In one method, the compound of formula II(b) is of the formula:

A specific compound of formula II(b) is (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate:

A particular crystalline form of this compound has a melting point of about 156° C. as measured by differential scanning calorimetry (DSC) (onset temperature). The form provides a X-ray powder diffraction (XRPD) pattern with peaks at one or more of about 7.7, 11.9, 12.4, 16.9, 19.5, 19.9, 21.9, 23.2, 24.1, and/or 27.7 degrees 2θ. As those skilled in the art are well aware, the relative intensities of peaks in a X-ray diffraction pattern of a crystalline form can vary depending on how the sample is prepared and how the data is collected. With this in mind, an example of a XRPD pattern of this crystalline form is provided in

wherein: each P2 is independently a hydroxyl protecting group stable under acidic conditions, or both P2s are taken together to provide a single protecting group; X′ is chlorine, bromine or iodine; and X″ is a leaving group (e.g., amino, alkoxyamino, hydroxy, halogen, alkoxy, phenoxy, carboxy, sulfoxy). In particular methods, each P2 is independently C(O)R6, or both P2s are taken together to provide C(R6)2, wherein each R6 is independently alkyl, aryl, alkylaryl, or arylalkyl.

and the salt of P1 is, for example, acyl chloride or acetic anhydride.


A particular crystalline form of this compound has a melting point of about 113° C. as measured by DSC (onset temperature). The form provides a XRPD pattern with peaks at one or more of about 7.6, 13.2, 17.0, 17.4, 18.6, 19.5, 20.5, 20.8 and/or 23.2 degrees 2θ. An example of a XRPD pattern of this crystalline form is provided in

under suitable conditions, wherein M is an appropriate metal, such as Na, K, Li, or Mg, X′ is Cl, Br, or I, and p is 0, 1, or 2, depending on the metal.

A particular crystalline form of this compound has a melting point of about 136° C. as measured by DSC (onset temperature). The form provides a XRPD pattern with peaks at one or more of about 9.0, 16.9, 17.6, 18.2, 18.4, 18.8 and/or 22.7 degrees 2θ. An example of a XRPD pattern of this crystalline form is provided in

Suitable reaction conditions are known in the art, and include those described below in the Examples. In general, L-(−)-xylose is cyclized under conditions sufficient to provide compound 1, which is then oxidized to provide compound 2, which is then contacted with morpholine under conditions sufficient to provide ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone. This invention encompasses compounds of formulae 1 and 2, including crystalline forms thereof.

A specific compound of formula IV is 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene:

A particular crystalline form of this compound has a melting point of about 65° C. (as determined by m.p. apparatus). The form provides a XRPD pattern with peaks at one or more of about 5.1, 13.5, 15.2, 20.3, 22.2 and/or 27.0 degrees 2θ. An example of a XRPD pattern of this crystalline form is provided in

More particular compounds are of the formula:

Specific compounds of formula IV(a) are (4-chloro-3-(4-ethoxybenzyl)phenyl)magnesium iodide and (4-chloro-3-(4-ethoxybenzyl)phenyl)magnesium chloride, shown below:







Claims (42)































Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/174,722 US8026347B2 (en) | 2007-07-26 | 2008-07-17 | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors |
| US13/207,576 US8293878B2 (en) | 2007-07-26 | 2011-08-11 | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US95212207P | 2007-07-26 | 2007-07-26 | |
| US12/174,722 US8026347B2 (en) | 2007-07-26 | 2008-07-17 | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/207,576 Continuation US8293878B2 (en) | 2007-07-26 | 2011-08-11 | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20090030198A1 US20090030198A1 (en) | 2009-01-29 |
| US8026347B2 true US8026347B2 (en) | 2011-09-27 |
Family
ID=39877996
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/174,722 Active 2030-03-13 US8026347B2 (en) | 2007-07-26 | 2008-07-17 | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors |
| US13/207,576 Active US8293878B2 (en) | 2007-07-26 | 2011-08-11 | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/207,576 Active US8293878B2 (en) | 2007-07-26 | 2011-08-11 | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors |
Country Status (25)
| Country | Link |
|---|---|
| US (2) | US8026347B2 (en) |
| EP (1) | EP2183263B1 (en) |
| JP (2) | JP5653213B2 (en) |
| KR (1) | KR101663324B1 (en) |
| CN (1) | CN101801989B (en) |
| AR (2) | AR067701A1 (en) |
| AT (1) | ATE530558T1 (en) |
| AU (1) | AU2008279424B2 (en) |
| BR (1) | BRPI0813840A2 (en) |
| CA (1) | CA2694029C (en) |
| CL (1) | CL2008002169A1 (en) |
| CO (1) | CO6260141A2 (en) |
| DK (1) | DK2183263T3 (en) |
| EA (1) | EA017411B1 (en) |
| EC (1) | ECSP109987A (en) |
| ES (1) | ES2375800T3 (en) |
| HK (1) | HK1143982A1 (en) |
| IL (1) | IL203209A (en) |
| NZ (1) | NZ582536A (en) |
| PL (1) | PL2183263T3 (en) |
| PT (1) | PT2183263E (en) |
| TW (2) | TWI419886B (en) |
| UA (1) | UA107175C2 (en) |
| WO (1) | WO2009014970A1 (en) |
| ZA (1) | ZA201000219B (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100016422A1 (en) * | 2008-07-17 | 2010-01-21 | Susan Margaret De Paul | Solid forms of (2s,3r,4r,5s,6r)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2h-pyran-3,4,5-triol and methods of their use |
| US20100311673A1 (en) * | 2006-09-29 | 2010-12-09 | Bryce Alden Harrison | Sulfanyl-tetrahydropyran-based compounds and methods of their use |
| US20110218159A1 (en) * | 2010-03-02 | 2011-09-08 | Philip Manton Brown | Methods of using inhibitors of sodium-glucose cotransporters 1 and 2 |
| US9394329B2 (en) | 2013-09-27 | 2016-07-19 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivatives and their uses in medicine |
Families Citing this family (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2730734C (en) | 2008-07-15 | 2017-04-25 | Theracos, Inc. | Deuterated 2,3,4-trihydroxy-tetrahydropyranyl-benzylbenzene compounds having sodium glucose cotransporter inhibitory activity |
| EP2334687B9 (en) | 2008-08-28 | 2012-08-08 | Pfizer Inc. | Dioxa-bicyclo[3.2.1.]octane-2,3,4-triol derivatives |
| PT2496583E (en) | 2009-11-02 | 2015-01-14 | Pfizer | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| BR112012011274A2 (en) | 2009-11-13 | 2016-04-12 | Astrazeneca Uk Ltd | reduced mass metformin formulation and its combination |
| ES2693686T3 (en) | 2009-11-13 | 2018-12-13 | Astrazeneca Ab | Immediate-release tablet formulations |
| HUE040486T2 (en) | 2009-11-13 | 2019-03-28 | Astrazeneca Ab | Bilayer tablet formulations |
| WO2011153712A1 (en) * | 2010-06-12 | 2011-12-15 | Theracos, Inc. | Crystalline form of benzylbenzene sglt2 inhibitor |
| WO2012031124A2 (en) | 2010-09-03 | 2012-03-08 | Bristol-Myers Squibb Company | Drug formulations using water soluble antioxidants |
| EP2670397B1 (en) | 2011-02-01 | 2020-05-13 | Bristol-Myers Squibb Company | Pharmaceutical formulations including an amine compound |
| US9034921B2 (en) | 2011-06-01 | 2015-05-19 | Green Cross Corporation | Diphenylmethane derivatives as SGLT2 inhibitors |
| EP2714052B1 (en) | 2011-06-03 | 2018-09-19 | Boehringer Ingelheim International GmbH | Sglt-2 inhibitors for treating metabolic disorders in patients treated with neuroleptic agents |
| US9193751B2 (en) | 2012-04-10 | 2015-11-24 | Theracos, Inc. | Process for the preparation of benzylbenzene SGLT2 inhibitors |
| CN108440614A (en) * | 2012-11-20 | 2018-08-24 | 莱西肯医药有限公司 | The inhibitor of sodium glucose cotransporter 1 |
| HUE064190T2 (en) | 2013-04-04 | 2024-03-28 | Boehringer Ingelheim Vetmedica Gmbh | Treatment of metabolic disorders in equine animals |
| CN105611920B (en) | 2013-10-12 | 2021-07-16 | 泰拉科斯萨普有限责任公司 | Preparation of hydroxy-diphenylmethane derivatives |
| CA3163455A1 (en) | 2013-12-17 | 2015-06-25 | Boehringer Ingelheim Vetmedica Gmbh | Treatment of metabolic disorders in feline animals |
| LT3485890T (en) | 2014-01-23 | 2023-07-25 | Boehringer Ingelheim Vetmedica Gmbh | Sglt2 inhibitors for treatment of metabolic disorders in canine animals |
| MX2016012705A (en) | 2014-04-01 | 2016-12-16 | Boehringer Ingelheim Vetmedica Gmbh | Treatment of metabolic disorders in equine animals. |
| EP3466958B1 (en) * | 2016-05-25 | 2020-04-29 | Crystal Pharmaceutical (Suzhou) Co., Ltd. | New crystal forms of sodium-glucose co-transporter inhibitor, processes for preparation and use thereof |
| WO2017206827A1 (en) * | 2016-05-28 | 2017-12-07 | 山东轩竹医药科技有限公司 | Crystal form of sodium-glucose cotransporter 2 inhibitor |
| CN113651803B (en) | 2016-06-17 | 2022-10-28 | 株式会社大熊制药 | Process for producing diphenylmethane derivative |
| WO2018073154A1 (en) | 2016-10-19 | 2018-04-26 | Boehringer Ingelheim International Gmbh | Combinations comprising an ssao/vap-1 inhibitor and a sglt2 inhibitor, uses thereof |
| CN107540685B (en) * | 2017-09-04 | 2020-05-05 | 杭州科巢生物科技有限公司 | Preparation method and intermediate of Sotagliflozin |
| JP2021520394A (en) | 2018-04-17 | 2021-08-19 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Pharmaceutical composition, treatment method and its use |
| CN108675976B (en) * | 2018-05-14 | 2020-07-14 | 浙江宏元药业股份有限公司 | 6-halogenated glucose carbon glycoside and preparation method and application thereof |
| CA3109107C (en) | 2018-08-13 | 2023-04-25 | Daewoong Pharmaceutical Co., Ltd. | Preparation of intermediate useful for synthesis of a diphenylmethane derivative for use as a sglt inhibitor |
| CN110818722B (en) * | 2018-08-14 | 2022-12-02 | 苏州鹏旭医药科技有限公司 | Three compounds, preparation method thereof and application thereof in synthesizing suogliflozin |
| UY38969A (en) | 2019-11-28 | 2021-05-31 | Boehringer Ingelheim Vetmedica Gmbh | USE OF SGLT-2 INHIBITORS IN DRYING NON-HUMAN MAMMALS |
| CN115087441A (en) | 2020-02-17 | 2022-09-20 | 勃林格殷格翰动物保健有限公司 | Use of SGLT-2 inhibitors for preventing and/or treating heart diseases in felines |
| WO2023006745A1 (en) | 2021-07-28 | 2023-02-02 | Boehringer Ingelheim Vetmedica Gmbh | Use of sglt-2 inhibitors for the prevention and/or treatment of hypertension in non-human mammals |
| WO2023006747A1 (en) | 2021-07-28 | 2023-02-02 | Boehringer Ingelheim Vetmedica Gmbh | Use of sglt-2 inhibitors for the prevention and/or treatment of renal diseases in non-human mammals |
| CN117715640A (en) | 2021-07-28 | 2024-03-15 | 勃林格殷格翰动物保健有限公司 | Use of SGLT-2 inhibitors for preventing and/or treating heart diseases in non-human mammals excluding felines, particularly canines |
| CN113880701A (en) * | 2021-10-10 | 2022-01-04 | 浙江司太立制药股份有限公司 | Antidiabetic drug intermediate and preparation method thereof |
| US20230381101A1 (en) | 2022-05-25 | 2023-11-30 | Boehringer Ingelheim Vetmedica Gmbh | Aqueous pharmaceutical compositions comprising sglt-2 inhibitors |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6414126B1 (en) | 1999-10-12 | 2002-07-02 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| US6515117B2 (en) | 1999-10-12 | 2003-02-04 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| US6555519B2 (en) | 2000-03-30 | 2003-04-29 | Bristol-Myers Squibb Company | O-glucosylated benzamide SGLT2 inhibitors and method |
| US6562791B1 (en) | 2002-03-29 | 2003-05-13 | Council Of Scientific And Industrial Research | Glucopyranoside, process for isolation thereof, pharmaceutical composition containing same and use thereof |
| US6683056B2 (en) | 2000-03-30 | 2004-01-27 | Bristol-Myers Squibb Company | O-aryl glucoside SGLT2 inhibitors and method |
| US6774112B2 (en) | 2001-04-11 | 2004-08-10 | Bristol-Myers Squibb Company | Amino acid complexes of C-aryl glucosides for treatment of diabetes and method |
| US6936590B2 (en) | 2001-03-13 | 2005-08-30 | Bristol Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| US7045665B2 (en) | 2000-03-17 | 2006-05-16 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| US7053060B2 (en) | 2000-11-30 | 2006-05-30 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| US7202350B2 (en) | 2003-03-14 | 2007-04-10 | Astellas Pharma Inc. | C-glycoside derivatives and salts thereof |
| US7250522B2 (en) | 2002-08-09 | 2007-07-31 | Taisho Pharmaceutical Co., Ltd. | Method for selective preparation of aryl 5-thio-β-D-aldohexopyranosides |
| US20100016422A1 (en) | 2008-07-17 | 2010-01-21 | Susan Margaret De Paul | Solid forms of (2s,3r,4r,5s,6r)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2h-pyran-3,4,5-triol and methods of their use |
| US7772378B2 (en) | 2005-02-23 | 2010-08-10 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted ((hetero)arylethynyl-benzyl)-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US7781577B2 (en) | 2006-09-29 | 2010-08-24 | Lexicon Pharmaceuticals, Inc. | Inhibitors of sodium glucose co-transporter 2 and methods of their use |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5753789A (en) * | 1996-07-26 | 1998-05-19 | Yale University | Oligonucleotides containing L-nucleosides |
| EP1495040A4 (en) * | 2002-04-12 | 2006-01-25 | Achillion Pharmaceuticals Inc | Method for synthesizing beta-l-fluoro-2',3'didehydcytidine (beta-l-fd4c) |
| AU2003299966A1 (en) | 2003-01-03 | 2004-08-10 | Bristol-Myers Squibb Company | Methods of producing c-aryl glucoside sglt2 inhibitors |
| US7368475B2 (en) * | 2003-03-12 | 2008-05-06 | Kemin Pharma Bvba | Furanose-type bicyclic carbohydrates with biological activity |
| WO2006018150A1 (en) * | 2004-08-11 | 2006-02-23 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituited cyclene, medicaments containing said compounds, use thereof and method for the production thereof |
| WO2008044268A1 (en) * | 2006-10-05 | 2008-04-17 | Panasonic Corporation | Transmitter apparatus and transmitting method |
| US7846945B2 (en) * | 2007-03-08 | 2010-12-07 | Lexicon Pharmaceuticals, Inc. | Piperdine-based inhibitors of sodium glucose co-transporter 2 and methods of their use |
-
2008
- 2008-07-17 AU AU2008279424A patent/AU2008279424B2/en active Active
- 2008-07-17 ES ES08826634T patent/ES2375800T3/en active Active
- 2008-07-17 JP JP2010518296A patent/JP5653213B2/en active Active
- 2008-07-17 DK DK08826634.1T patent/DK2183263T3/en active
- 2008-07-17 US US12/174,722 patent/US8026347B2/en active Active
- 2008-07-17 KR KR1020107001659A patent/KR101663324B1/en active IP Right Grant
- 2008-07-17 UA UAA201002095A patent/UA107175C2/en unknown
- 2008-07-17 PL PL08826634T patent/PL2183263T3/en unknown
- 2008-07-17 NZ NZ582536A patent/NZ582536A/en unknown
- 2008-07-17 WO PCT/US2008/070250 patent/WO2009014970A1/en active Application Filing
- 2008-07-17 BR BRPI0813840A patent/BRPI0813840A2/en not_active Application Discontinuation
- 2008-07-17 EA EA201070186A patent/EA017411B1/en not_active IP Right Cessation
- 2008-07-17 PT PT08826634T patent/PT2183263E/en unknown
- 2008-07-17 EP EP08826634A patent/EP2183263B1/en active Active
- 2008-07-17 CN CN200880100489.2A patent/CN101801989B/en active Active
- 2008-07-17 AT AT08826634T patent/ATE530558T1/en active
- 2008-07-17 CA CA2694029A patent/CA2694029C/en active Active
- 2008-07-21 TW TW097127665A patent/TWI419886B/en active
- 2008-07-21 TW TW102129521A patent/TWI506024B/en active
- 2008-07-24 CL CL2008002169A patent/CL2008002169A1/en unknown
- 2008-07-25 AR ARP080103246A patent/AR067701A1/en active IP Right Grant
-
2010
- 2010-01-10 IL IL203209A patent/IL203209A/en active IP Right Grant
- 2010-01-12 ZA ZA2010/00219A patent/ZA201000219B/en unknown
- 2010-02-23 EC EC2010009987A patent/ECSP109987A/en unknown
- 2010-02-25 CO CO10022864A patent/CO6260141A2/en not_active Application Discontinuation
- 2010-11-12 HK HK10110567.0A patent/HK1143982A1/en unknown
-
2011
- 2011-08-11 US US13/207,576 patent/US8293878B2/en active Active
-
2013
- 2013-08-28 JP JP2013176646A patent/JP5764174B2/en active Active
-
2018
- 2018-08-03 AR ARP180102213 patent/AR112669A2/en active IP Right Grant
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6515117B2 (en) | 1999-10-12 | 2003-02-04 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| US6414126B1 (en) | 1999-10-12 | 2002-07-02 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| US7045665B2 (en) | 2000-03-17 | 2006-05-16 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| US6555519B2 (en) | 2000-03-30 | 2003-04-29 | Bristol-Myers Squibb Company | O-glucosylated benzamide SGLT2 inhibitors and method |
| US6683056B2 (en) | 2000-03-30 | 2004-01-27 | Bristol-Myers Squibb Company | O-aryl glucoside SGLT2 inhibitors and method |
| US7053060B2 (en) | 2000-11-30 | 2006-05-30 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxybenzylbenzene derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| US6936590B2 (en) | 2001-03-13 | 2005-08-30 | Bristol Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| US6774112B2 (en) | 2001-04-11 | 2004-08-10 | Bristol-Myers Squibb Company | Amino acid complexes of C-aryl glucosides for treatment of diabetes and method |
| US6562791B1 (en) | 2002-03-29 | 2003-05-13 | Council Of Scientific And Industrial Research | Glucopyranoside, process for isolation thereof, pharmaceutical composition containing same and use thereof |
| US7250522B2 (en) | 2002-08-09 | 2007-07-31 | Taisho Pharmaceutical Co., Ltd. | Method for selective preparation of aryl 5-thio-β-D-aldohexopyranosides |
| US7202350B2 (en) | 2003-03-14 | 2007-04-10 | Astellas Pharma Inc. | C-glycoside derivatives and salts thereof |
| US7772378B2 (en) | 2005-02-23 | 2010-08-10 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted ((hetero)arylethynyl-benzyl)-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US7781577B2 (en) | 2006-09-29 | 2010-08-24 | Lexicon Pharmaceuticals, Inc. | Inhibitors of sodium glucose co-transporter 2 and methods of their use |
| US20100016422A1 (en) | 2008-07-17 | 2010-01-21 | Susan Margaret De Paul | Solid forms of (2s,3r,4r,5s,6r)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2h-pyran-3,4,5-triol and methods of their use |
Non-Patent Citations (4)
| Title |
|---|
| Davis, N.J. and Flitsch, S.L., Tetrahedron Letters 34(7):1181-1184 (1993). |
| Handlon, A.L., Expert Opin Therapeutic Patents 15(11):1531-1540 (2005). |
| Kanazawa et al. Polymer (2006), vol. 47, pp. 176-183. * |
| Search Report and Written Opinion for Corresponding International Application PCT/US2008/070250, dated Nov. 19, 2008. |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100311673A1 (en) * | 2006-09-29 | 2010-12-09 | Bryce Alden Harrison | Sulfanyl-tetrahydropyran-based compounds and methods of their use |
| US8476413B2 (en) * | 2006-09-29 | 2013-07-02 | Lexicon Pharmaceuticals, Inc. | Sulfanyl-tetrahydropyran-based compounds and methods of their use |
| US20190099436A1 (en) * | 2006-09-29 | 2019-04-04 | Lexicon Pharmaceuticals, Inc. | Sodium glucose co-transporter inhibitors and methods of their use |
| US20100016422A1 (en) * | 2008-07-17 | 2010-01-21 | Susan Margaret De Paul | Solid forms of (2s,3r,4r,5s,6r)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2h-pyran-3,4,5-triol and methods of their use |
| US8217156B2 (en) * | 2008-07-17 | 2012-07-10 | Lexicon Pharmaceuticals, Inc. | Solid forms of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol and methods of their use |
| US9067962B2 (en) | 2008-07-17 | 2015-06-30 | Lexicon Pharmaceuticals, Inc. | Methods of treating diabetes |
| US20110218159A1 (en) * | 2010-03-02 | 2011-09-08 | Philip Manton Brown | Methods of using inhibitors of sodium-glucose cotransporters 1 and 2 |
| US9394329B2 (en) | 2013-09-27 | 2016-07-19 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivatives and their uses in medicine |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8026347B2 (en) | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors | |
| JP6283337B2 (en) | (2S, 3R, 4R, 5S, 6R) -2- (4-Chloro-3- (4-ethoxybenzyl) phenyl) -6- (methylthio) tetrahydro-2H-pyran-3,4,5-triol Form and method of use | |
| US20190099436A1 (en) | Sodium glucose co-transporter inhibitors and methods of their use | |
| US7846945B2 (en) | Piperdine-based inhibitors of sodium glucose co-transporter 2 and methods of their use | |
| AU2013206276A1 (en) | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors | |
| Jacobsen | Dioxolanylium ions derived from carbohydrates. X: Nucleophilic trans opening with the trichloroacetimidoyl neighbouring group |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: LEXICON PHARMACEUTICALS, INC., TEXAS Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:GOODWIN, NICOLE C.;HARRISON, BRYCE A.;IIMURA, SHINYA;AND OTHERS;REEL/FRAME:021409/0960;SIGNING DATES FROM 20080804 TO 20080806 Owner name: LEXICON PHARMACEUTICALS, INC., TEXAS Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:GOODWIN, NICOLE C.;HARRISON, BRYCE A.;IIMURA, SHINYA;AND OTHERS;SIGNING DATES FROM 20080804 TO 20080806;REEL/FRAME:021409/0960 |
|
| STCF | Information on status: patent grant |
Free format text: PATENTED CASE |
|
| FPAY | Fee payment |
Year of fee payment: 4 |
|
| AS | Assignment |
Owner name: BIOPHARMA CREDIT PLC, UNITED KINGDOM Free format text: SECURITY INTEREST;ASSIGNOR:LEXICON PHARMACEUTICALS, INC.;REEL/FRAME:044958/0377 Effective date: 20171204 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 8TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1552); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 8 |
|
| AS | Assignment |
Owner name: LEXICON PHARMACEUTICALS, INC., TEXAS Free format text: RELEASE BY SECURED PARTY;ASSIGNOR:BIOPHARMA CREDIT PLC;REEL/FRAME:053767/0445 Effective date: 20200908 |
|
| AS | Assignment |
Owner name: OXFORD FINANCE LLC, AS COLLATERAL AGENT, VIRGINIA Free format text: SECURITY INTEREST;ASSIGNOR:LEXICON PHARMACEUTICALS, INC.;REEL/FRAME:059432/0709 Effective date: 20220317 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 12TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1553); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 12 |